El-MAVEN User Interface¶
Following is the general workflow involved in El-MAVEN:
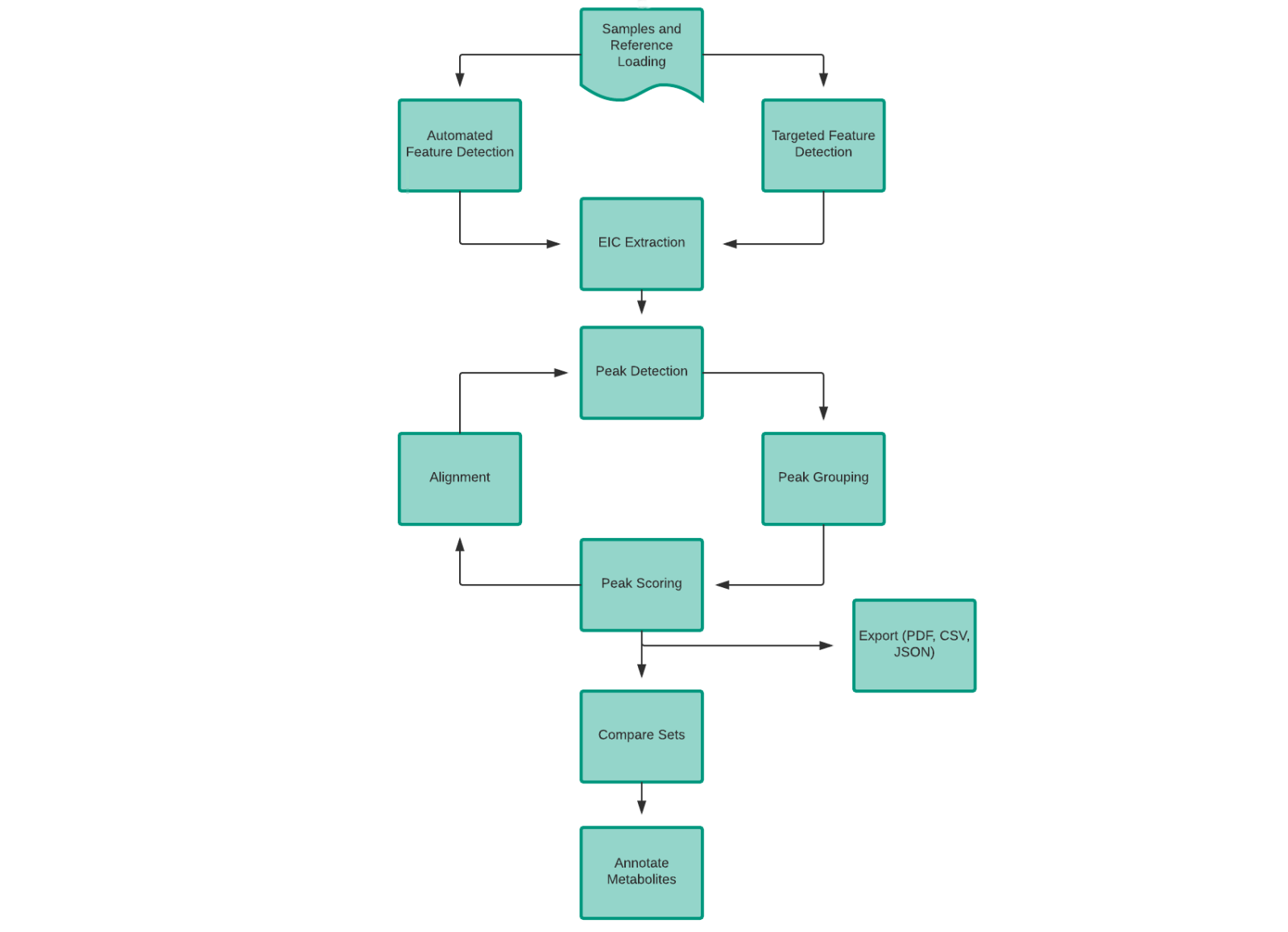
Peak detection, alignment, grouping and scoring are done multiple times for best results in the El-MAVEN workflow. Data from different cohorts can be compared using visualisation tools and easily exported to other formats.
El-MAVEN User Interface¶
Global Settings¶
Global Settings can be changed from the Options dialog  .
.
Instrumentation
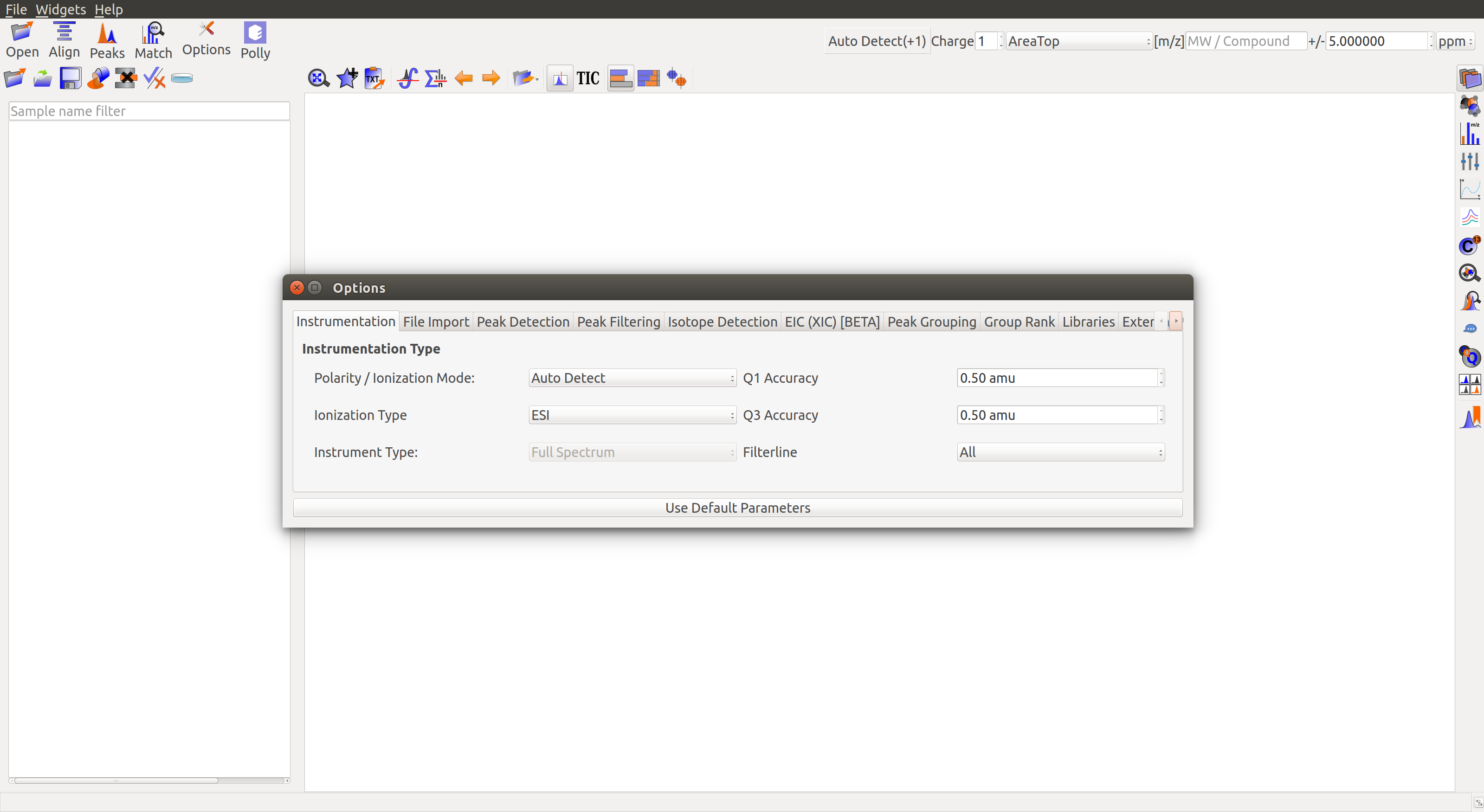
- Polarity/Ionization mode: Polarity information is required for m/z calculation. Users can set the polarity of the metabolites in their experiment from the drop-down list or set it to Auto-detect.
- Ionization type: Ionization methods can affect m/z calculation. Drop-down provides a list of the most popular ionization types.
- Q1 accuracy: This is the mass resolution in amu of the first quadrapole.
- Q3 accuracy: This is the mass resolution in amu of the third quadrapole.
- Filterline: The drop-down lists different mass ranges and allows the user to process the data in these ranges separately with different parameters. Primarily used for polarity-switching experiments.
File Import
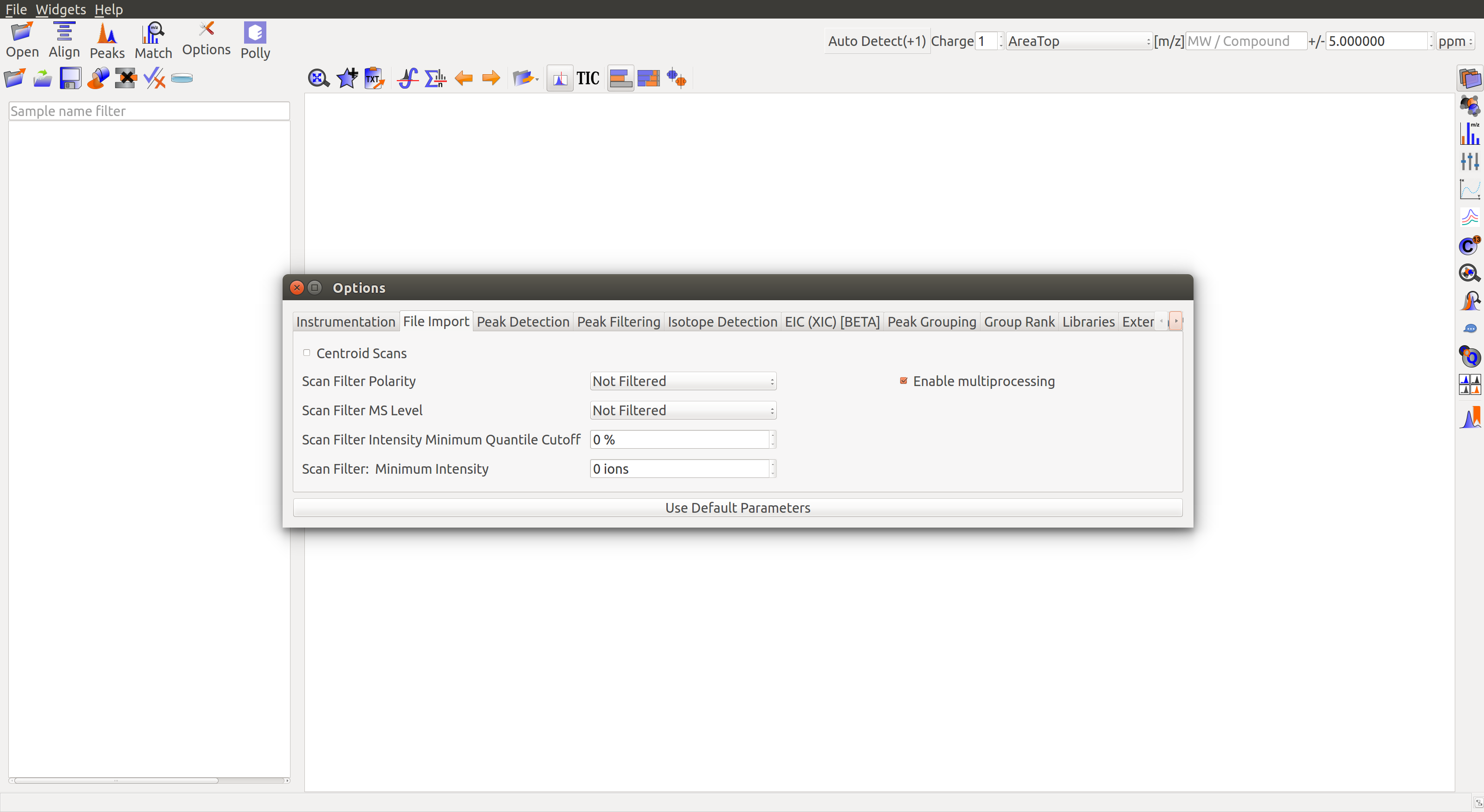
- Centroid Scans: Centroid acquisition is an acquisition method where only centroid m/z and intensity are stored. Centroid m/z is calculated based on the average m/z value weighted by the intensity and m/z values are assigned based on a calibration file. Users may leave the box unchecked if they have the centroid data or check the box if centroiding has to be done in El-MAVEN.
- Scan Filter Polarity: Users may choose to import scans based on the polarity of ions in the scan. Especially helpful in polarity-switching experiments.
- Scan Filter MS Level: Users may choose to import only MS1 or MS2 scans. This feature can be used with MS/MS data.
- Scan Filter Minimum Intensity: Sets a minimum threshold for reading in intensity values.
- Scan Filter Intensity Minimum Quantile Cutoff: Scans with x% of their intensity values below the threshold will be filtered out during import.
- Enable Multiprocessing: In order to reduce the sample load time, El-MAVEN uses multiprocessing. This behavior can be changed by the user.
Peak Detection
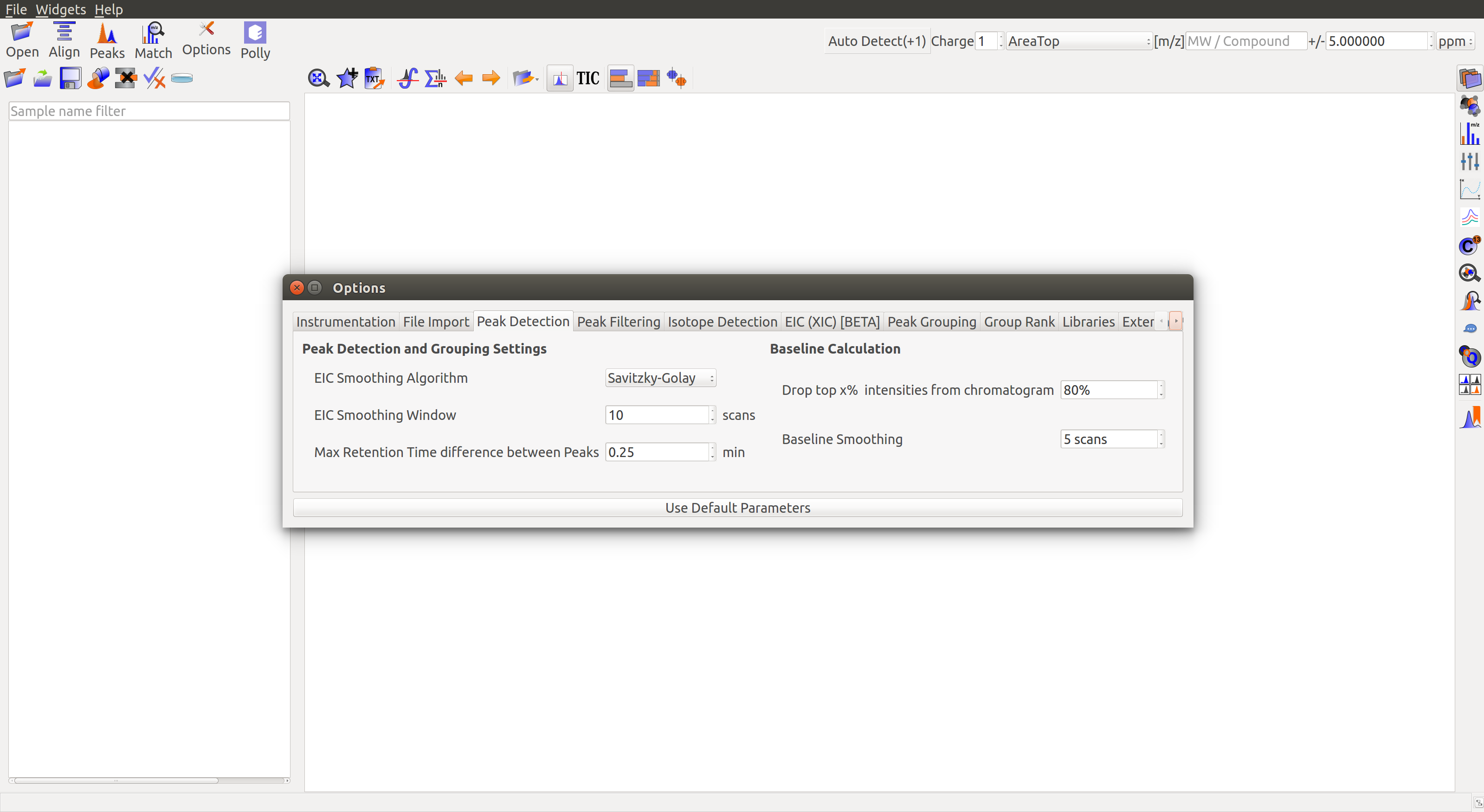
Peak Grouping and Grouping Settings
EIC Smoothing Algorithm: Smoothing of data points helps in increasing the signal/noise ratio. There are three algorithms provided for EIC smoothing:
- Savitzky-Golay: It preserves the original shape and features of the signal better than most other filters (Learn more).
- Gaussian: It reduces noise by averaging over the neighborhood with the central pixel having higher weight but successfully preserves sharp edges. (Learn more).
- Moving Average: It takes the simple average of all points over time. Signal behavior is not natural. Least preferred method for smoothing (Learn more).
EIC Smoothing Window: Number of scans used for fitting in the smoothing algorithm can be adjusted here.
Maximum Retention Time Difference Between Peaks: Set a limit to retention time (RT) difference between peaks in a group. Increase the value if alignment fails to center peaks satisfactorily.
Baseline Calculation
- Drop top x% intensities from chromatogram: Set the baseline for every peak. Baseline is obtained once x% of the highest intensities in a peak are removed from consideration. Baseline should be set high when there is more noise in the data.
- Baseline Smoothing: Number of scans used for fitting in the smoothing algorithm can be adjusted here.
Peak Filtering
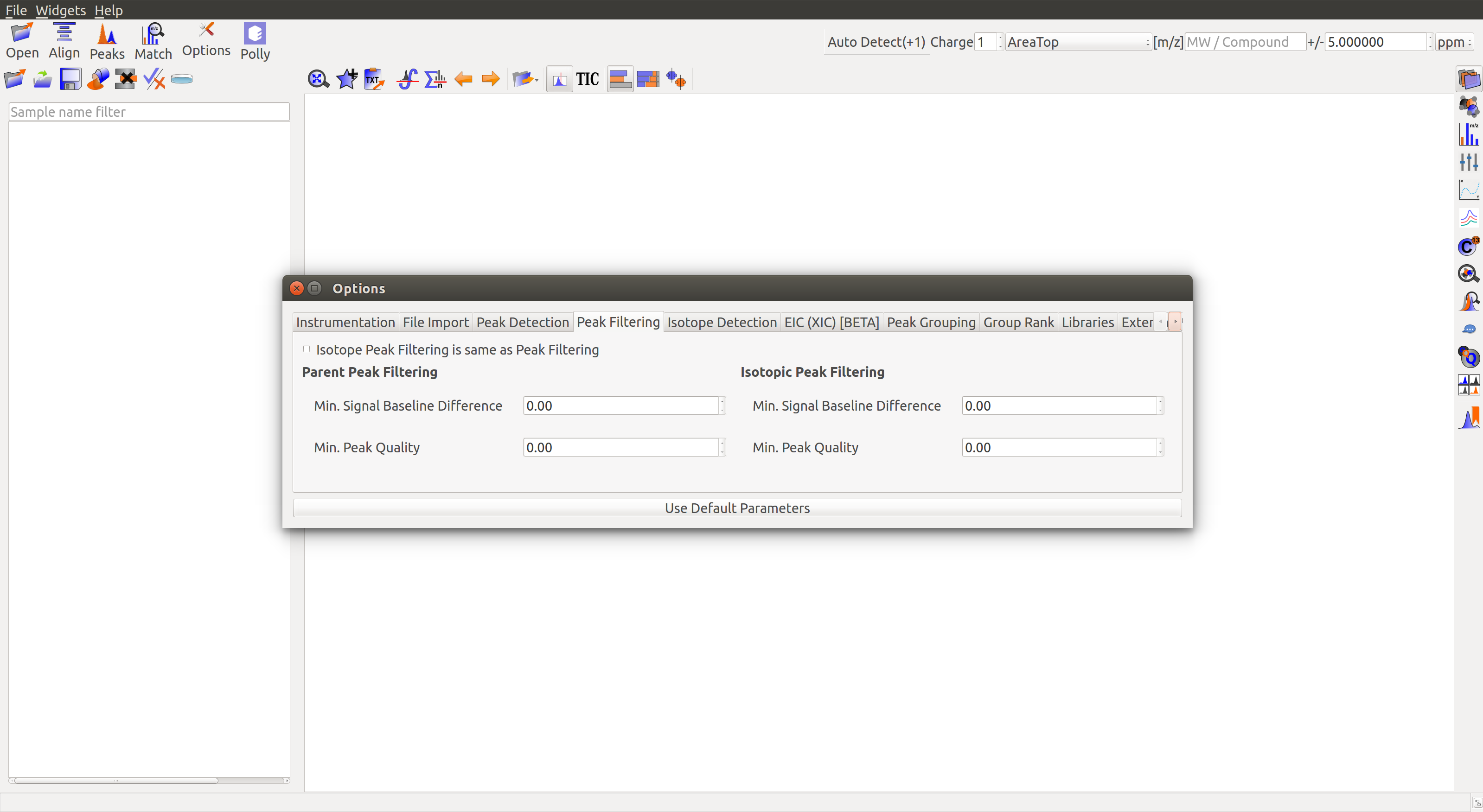
- Minimum Signal Baseline Difference: Set the minimum difference between intensity and baseline to detect any signal as a valid peak.
Isotope Detection¶
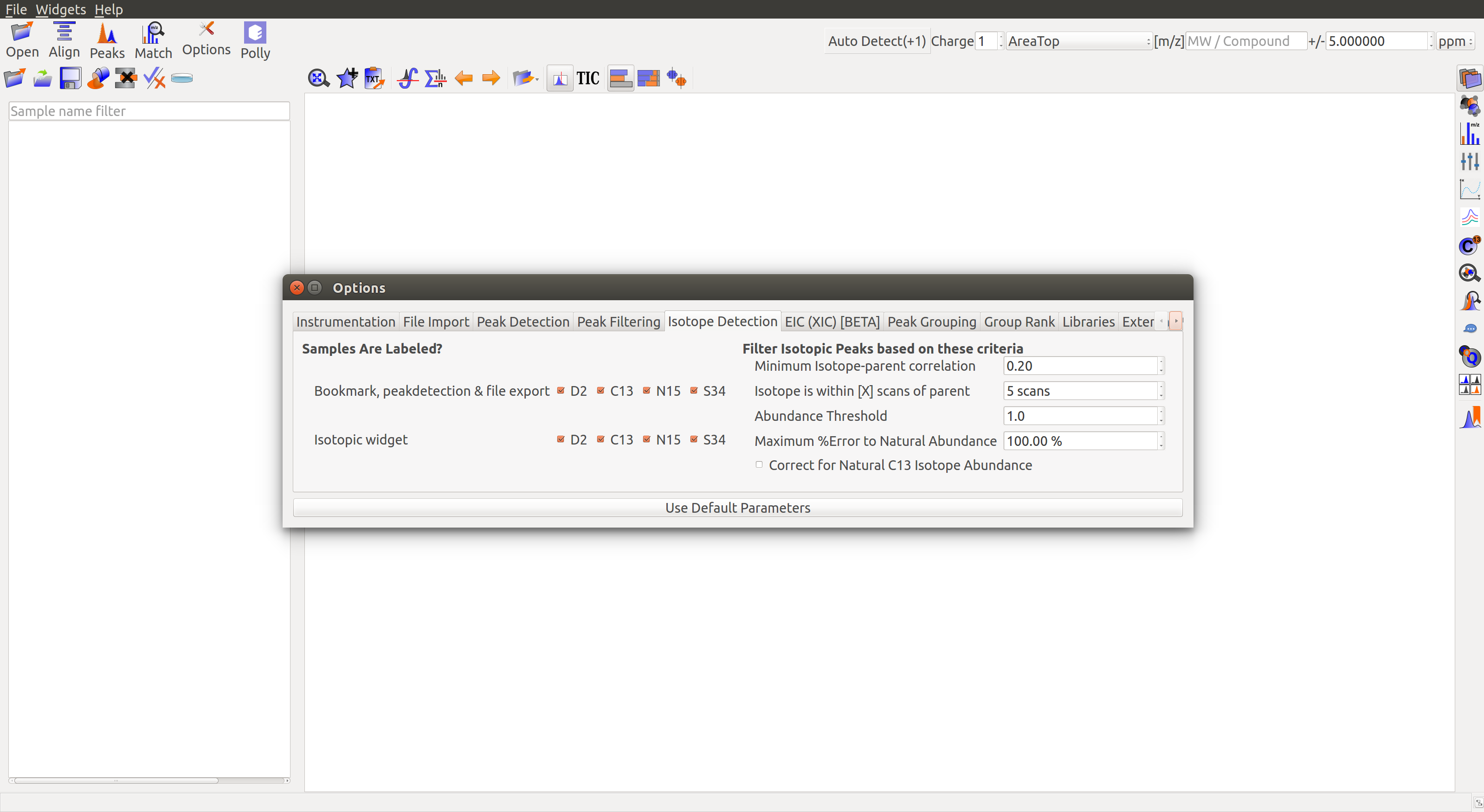
Are Samples Labeled?
- Bookmarks, peak detection, file export: Select the labeled atoms that should be used in bookmarking, peak detection and export. D2: Deuterium, C13: Labeled carbon, N15: Labeled nitrogen, S34: Labeled sulphur.
- Isotopic widget: Select the labeled atoms that should be displayed in the isotopic widget. D2: Deuterium, C13: Labeled carbon, N15: Labeled nitrogen, S34: Labeled sulphur.
- Number of M+n isotopes: Set the maximum number of labeled atoms per ion in the experiment.
- Abundance Threshold: Set the minimum threshold for isotopic abundance. Isotopic abundance is the ratio of intensity of isotopic peak over the parent peak.
Filter Isotopic Peaks based on these criteria
- Minimum Isotope-Parent Correlation: Set the minimum threshold for isotope-parent peak correlation. This correlation is a measure of how often they appear together.
- Isotope is within [X] scans of parent: Set the maximum scan difference between isotopic and parent peaks. This is a measure of how closely they appear together on the retention time scale.
- Maximum % Error to Natural Abundance: Set the maximum natural abundance error expected. Natural abundance of an isotope is the expected ratio of amount of isotope over the amount of parent molecule in nature. Error is the difference between observed and natural abundance as a fraction of natural abundance.
- Correct for Natural C13 Isotope Abundance: Check the box to correct for natural C13 abundance.
EIC (XIC) [BETA]
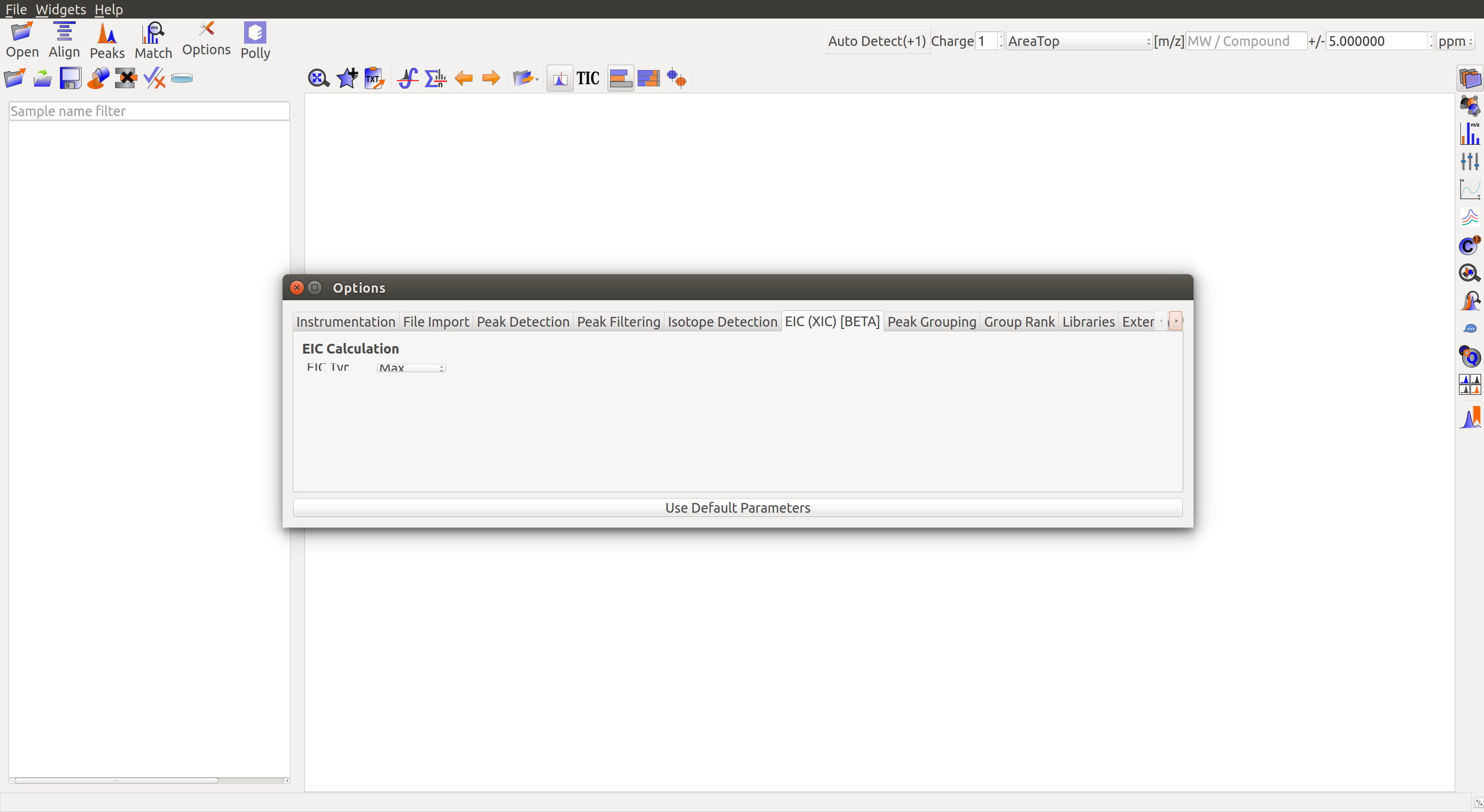
EIC Type: Select a method to merge EICs over m/z. There are two options:
- MAX: Merged EIC is created by taking the maximum intensity across the m/z window at a particular scan.
- SUM: Merged EIC is created by taking the sum average of intensities across the m/z window at a particular scan.
Peak Grouping
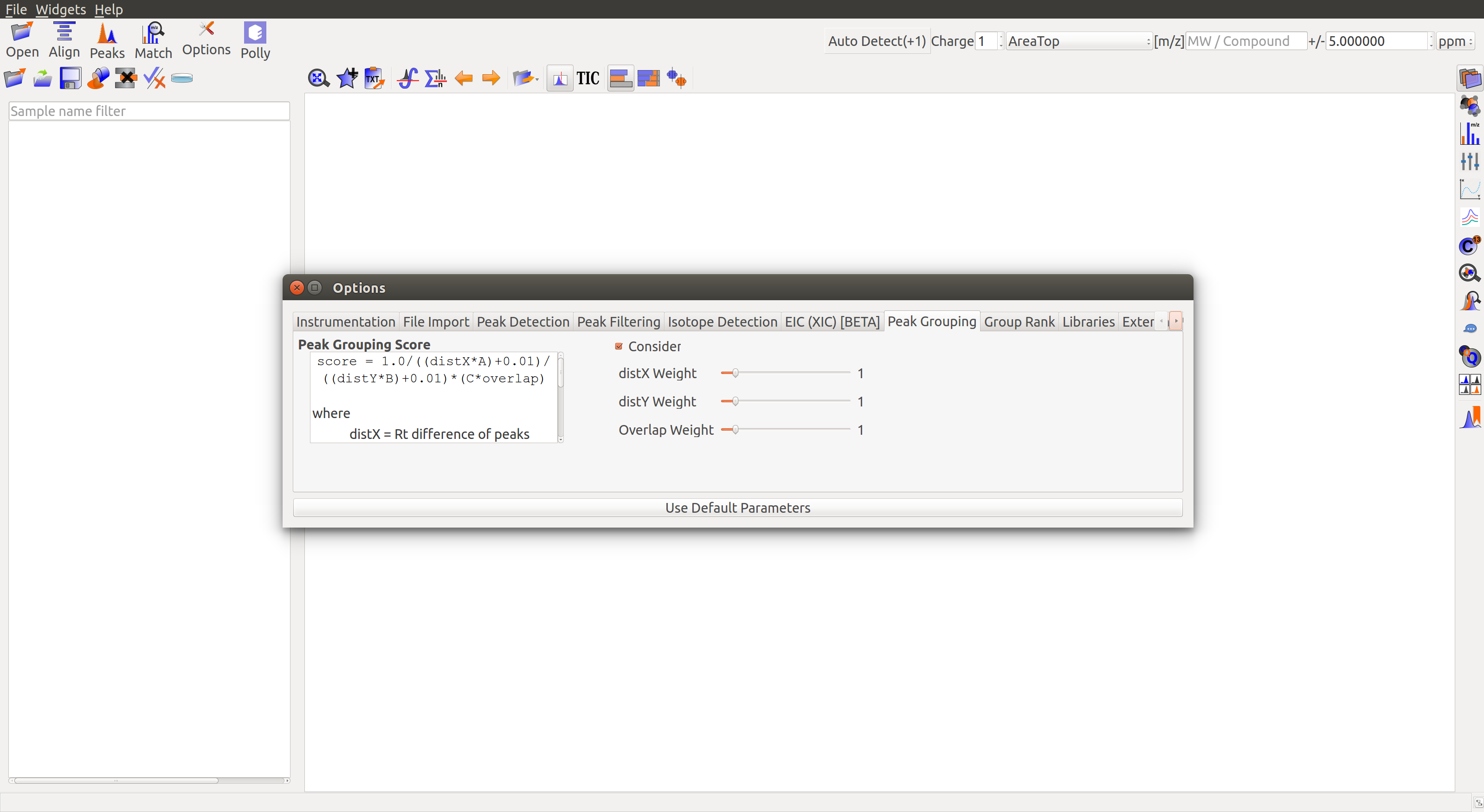
Peak Grouping Score: Peaks are assigned a grouping score to determine whether they should be grouped together. There are two formulas for grouping score calculation:
- score = 1.0/((distX * A) + 0.01)/((distY * B) + 0.01) * (C * overlap)
- score = 1.0/((distX * A) + 0.01)/((distY * B) + 0.01)
The score depends on the following 3 parameters and their weights:
RT difference or DistX: Difference in retention time between the peaks under comparison. Closer peaks are assigned a higher score.
Intensity difference or DistY: Difference in intensity between peaks under comparison. Smaller difference accounts for a higher score.
Overlap: Fraction of retention time overlap between the peaks under comparison. Greater overlap accounts for a higher score.
- Uncheck Consider Overlap to calculate grouping score without overlap.
- Sliders are provided to adjust the weights attached to each of the three parameters.
Group Rank
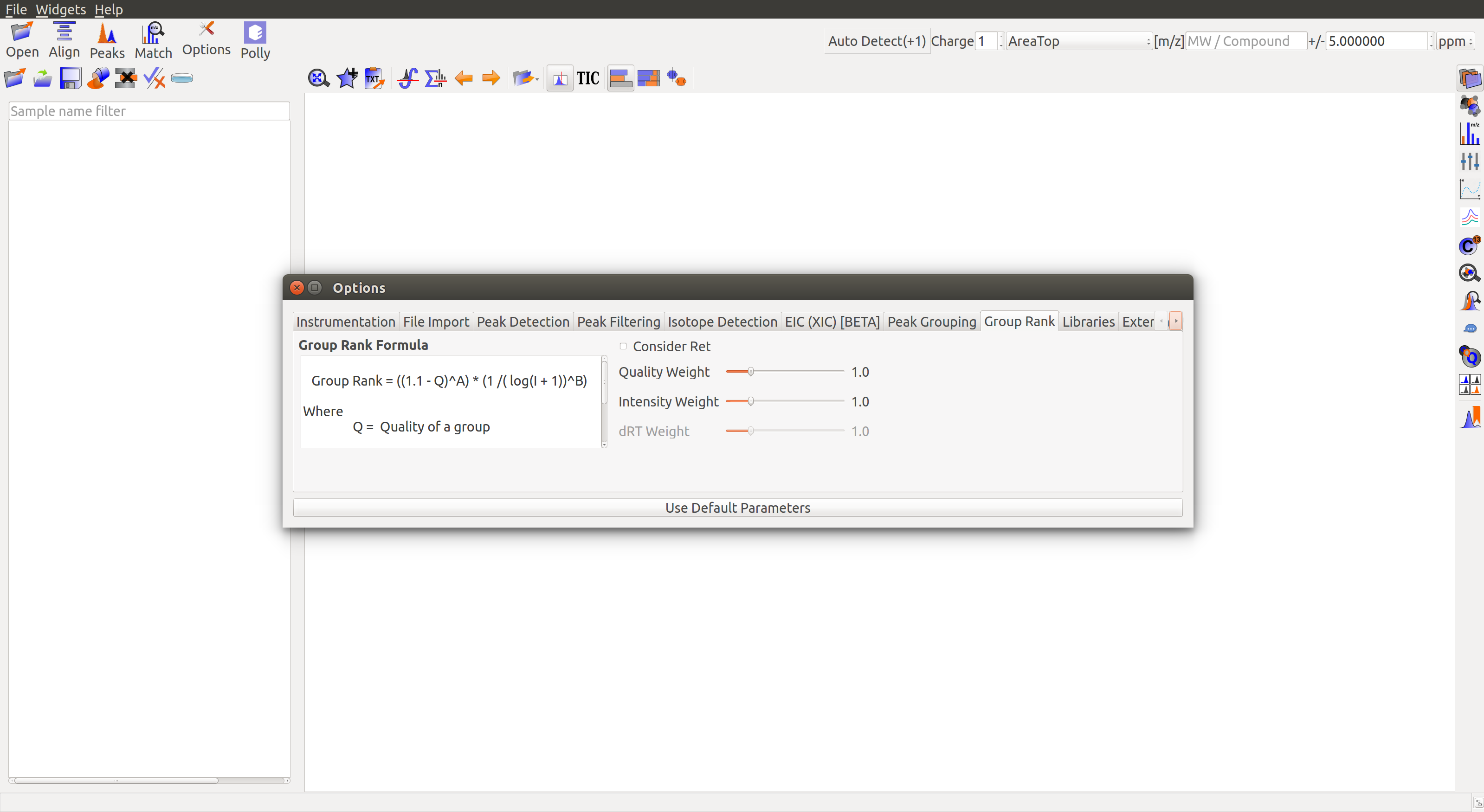
Group Rank Formula: Group rank is one of the parameters for group filtering. There are two formulas below for group rank calculation:
- Group Rank = ((1.1 - Q) ^ A) * (1/(log(I + 1)) ^ B)
- Group Rank = ((1.1 - Q) ^ A) * (1/(log(I + 1)) ^ B) * (dRT) ^ (2 * C)
The score depends on the following 3 parameters and their respective weights A, B and C:
Q or Group Quality: Maximum peak quality of a group. Peaks are assigned a quality score by a machine learning algorithm in El-MAVEN. Better quality leads to a higher rank.
I or Group Intensity: Maximum intensity of a group. Better intensity leads to a higher rank.
dRT or RT difference: Difference between expected retention time and group mean RT.
- Consider Retention Time: Check the box to use formula (b) for group rank calculation. Formula (a) is used by default.
- Quality Weight: Adjust slider to set weight for group quality in group rank calculation.
- Intensity Weight: Adjust slider to set weight for group intensity in group rank calculation.
- dRT Weight: Adjust slider to set weight for retention time difference in group rank calculation. The slider is disabled if Consider Retention Time is unchecked.
Sample Upload¶
Load Sample Files
Load  sample files into El-MAVEN and click on Show Samples Widget
sample files into El-MAVEN and click on Show Samples Widget  on the widget bar to show/hide the project space. Blanks will not show up in the sample list if the file names start with ‘blan’ or ‘blank’.
on the widget bar to show/hide the project space. Blanks will not show up in the sample list if the file names start with ‘blan’ or ‘blank’.
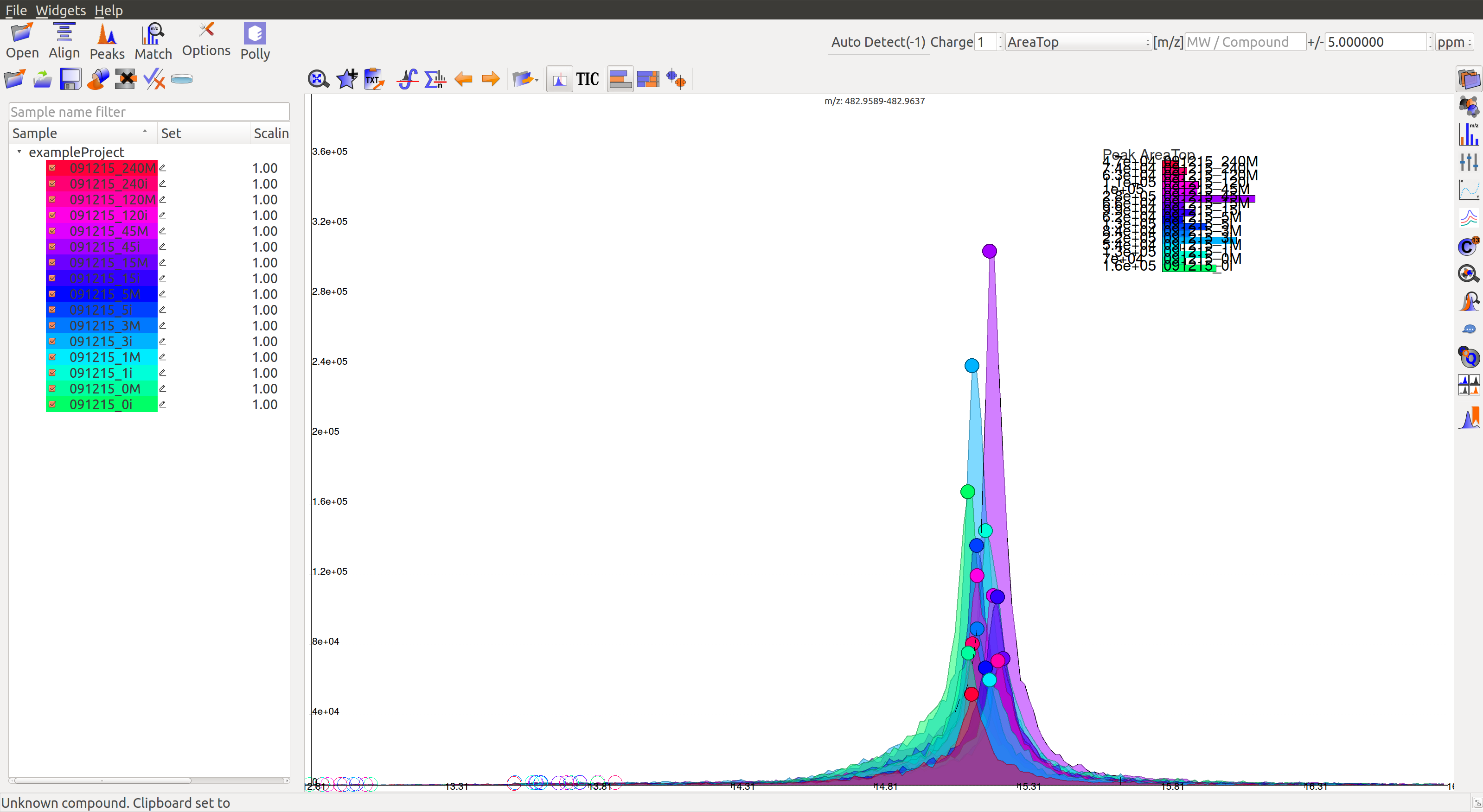
Load sample files into El-MAVEN and click on Show Samples Widget on the widget bar to show/hide the project space. Blanks will not show up in the sample list if the file names start with ‘blan’ or ‘blank’.
There are three columns in the project space:
- Sample: This column has the sample name and the random color assigned to the sample. Double-click the sample name to change the color.
- Set: The set column holds the cohort name for every sample. For example: subjects and controls.
- Scaling: This column holds the normalization constant for every sample. For example, all intensities in a sample will be halved if the constant is two. This is done to normalize data if sample volumes are different.
Sample Space Menu
Load Project: Sample files can be loaded here.
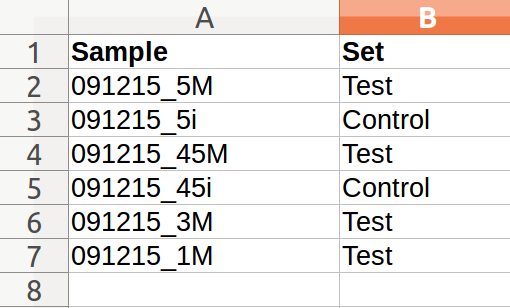
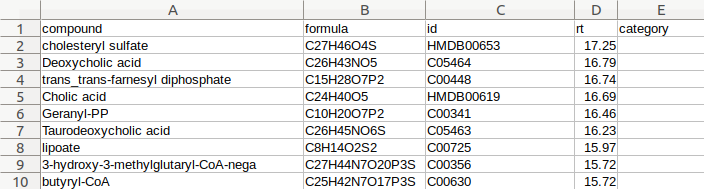
Load Meta: Users may upload a meta file with sample and set names in a comma separated file (.csv) or double-click to enter text. Meta file template is shown below:
Save Project as: Current state of El-MAVEN can be saved in a .mzroll file for future use. All the settings, EICs and peak tables are stored in the file and may be reloaded at any point in the future.
Change Sample Color: Sample colors can be changed by either clicking on this menu button or double-clicking the sample name. Users can pick a color of their choice to represent their samples.
Remove Samples: Apart from deselecting samples, users also have the option to remove samples from the project space. The sample files will not be deleted, only removed from El-MAVEN’s project space.
Show/Hide Selected Samples: Samples can be selected/deselected in batches. This is especially helpful when dealing with large datasets as the EIC window gets increasingly noisy with more samples.
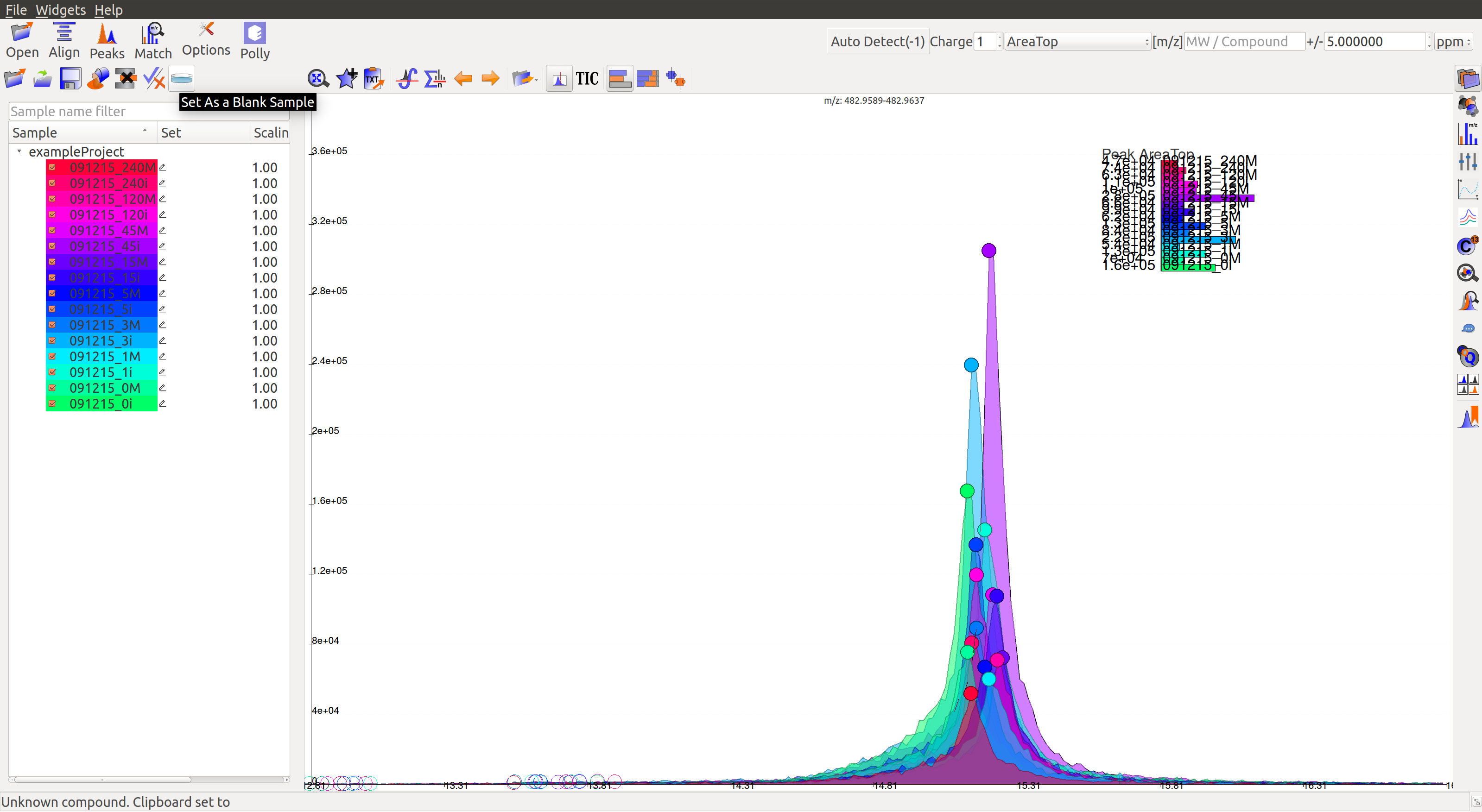
Mark Sample as Blank: Users can select sample files and set them as blanks as depicted below. Clicking the button again will reverse the move.
Compound Database¶
Load Reference File
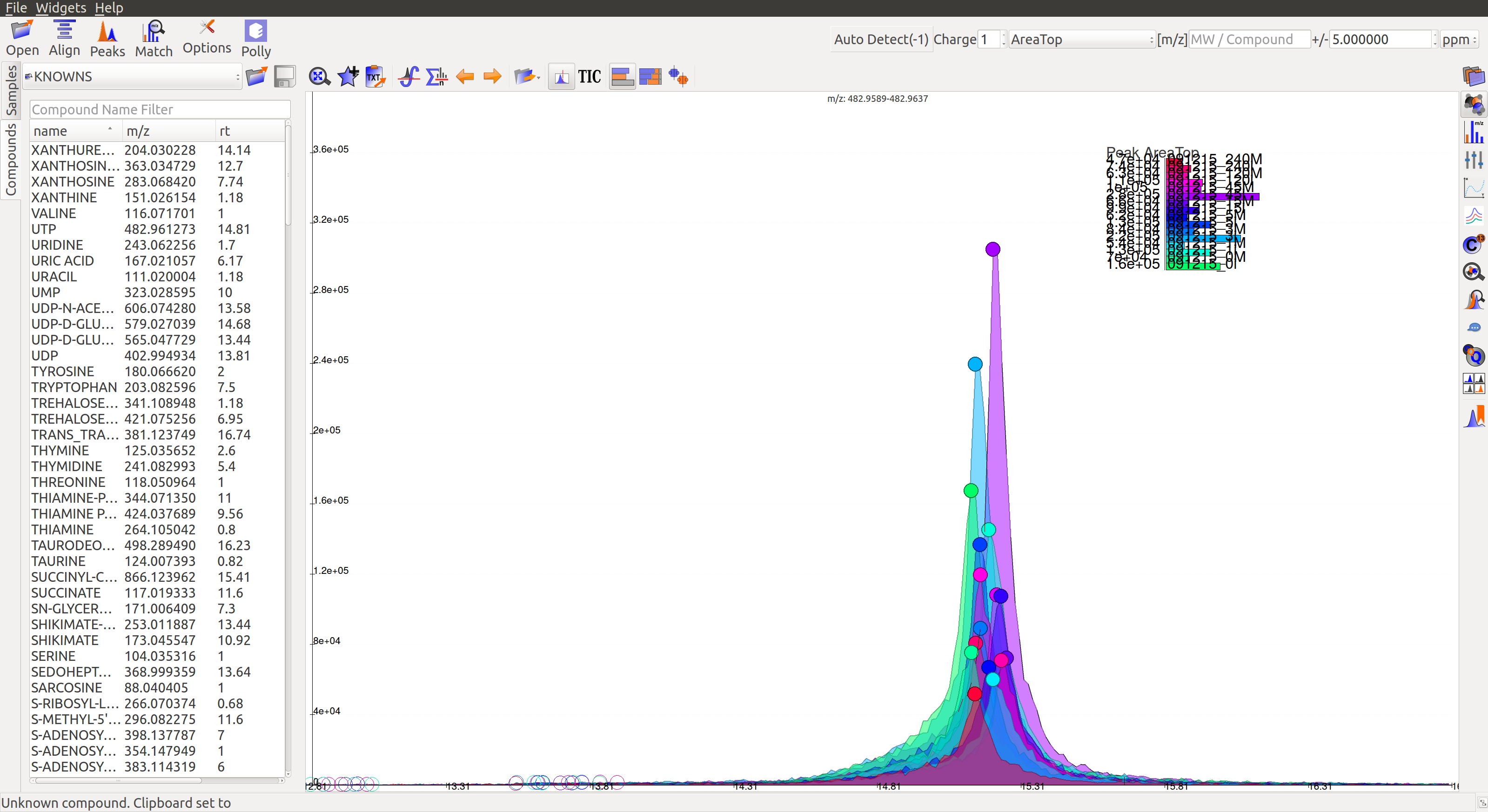
Reference file contains a list of metabolites and their properties that are used for peak detection. This is a comma separated (.csv) or tab separated (.tab) file with compound name, id, formula, mass, expected retention time and category. It is preferable but not necessary to have retention time information in the reference file but either mass or formula is required. In case both mass and formula are provided, formula will be used to calculate the m/z. Click on the Show Compounds Widget on the widget toolbar to view the compounds panel. Users may upload a new reference file or use any of the default files loaded on start-up.
EIC¶
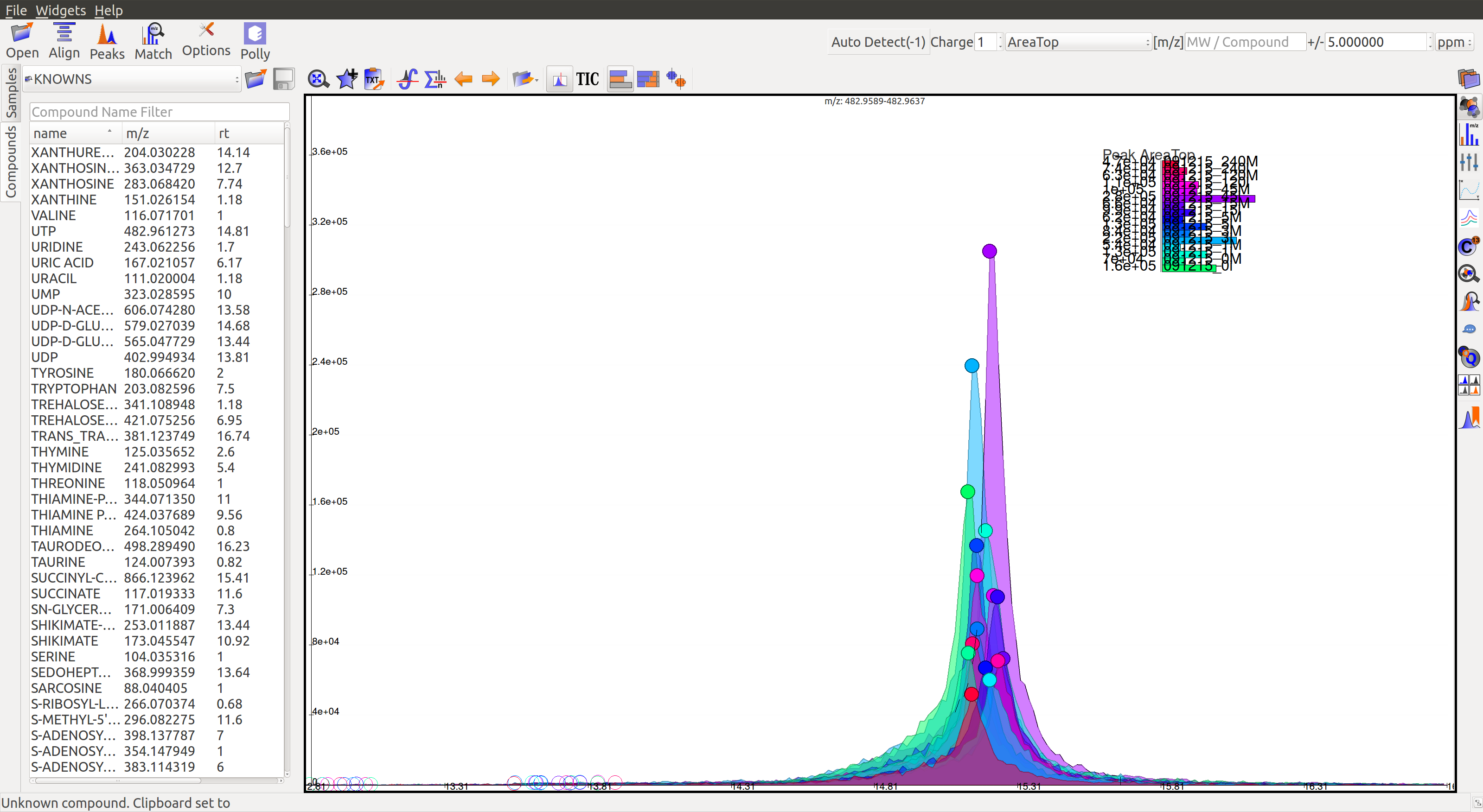
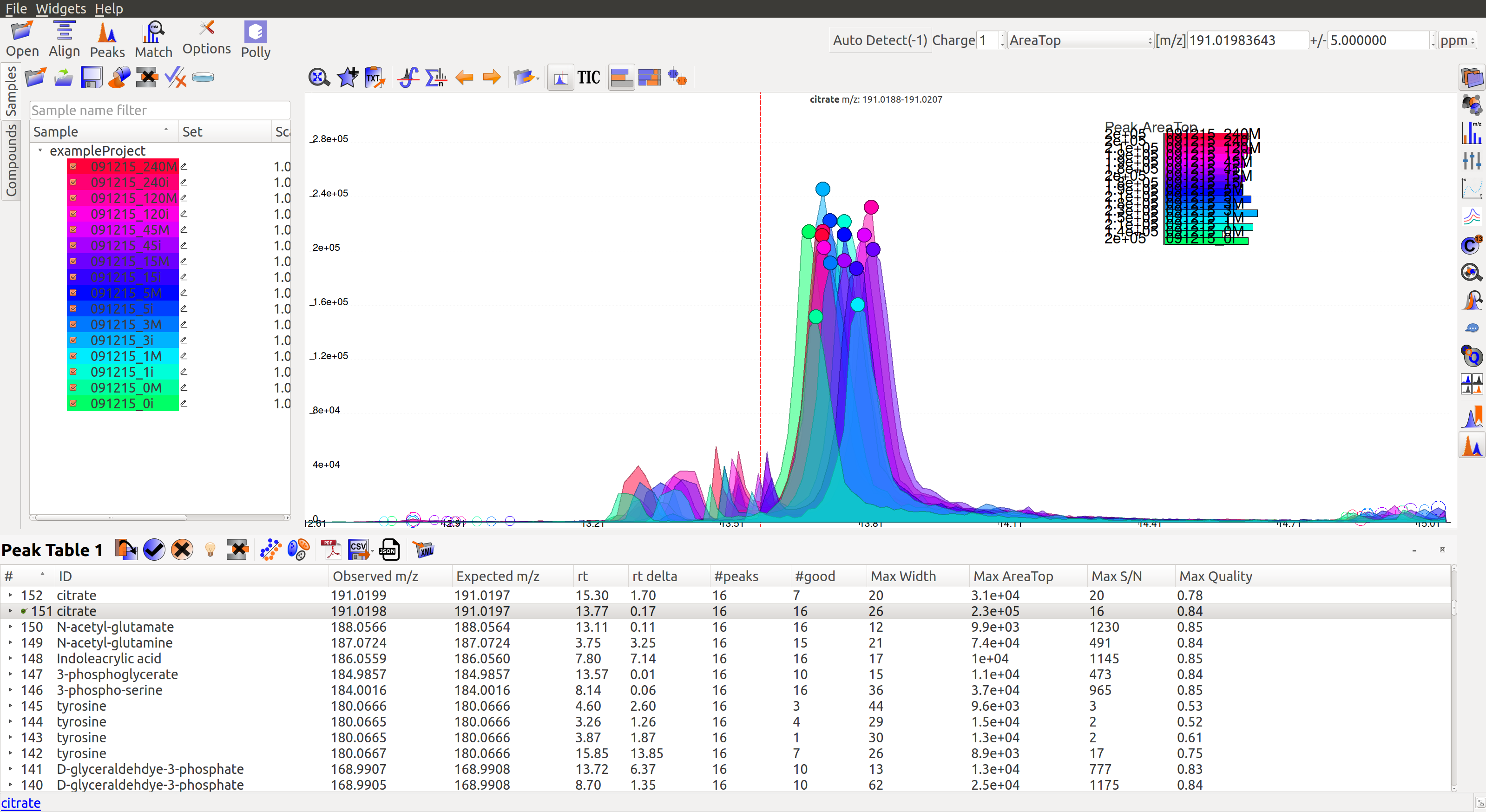
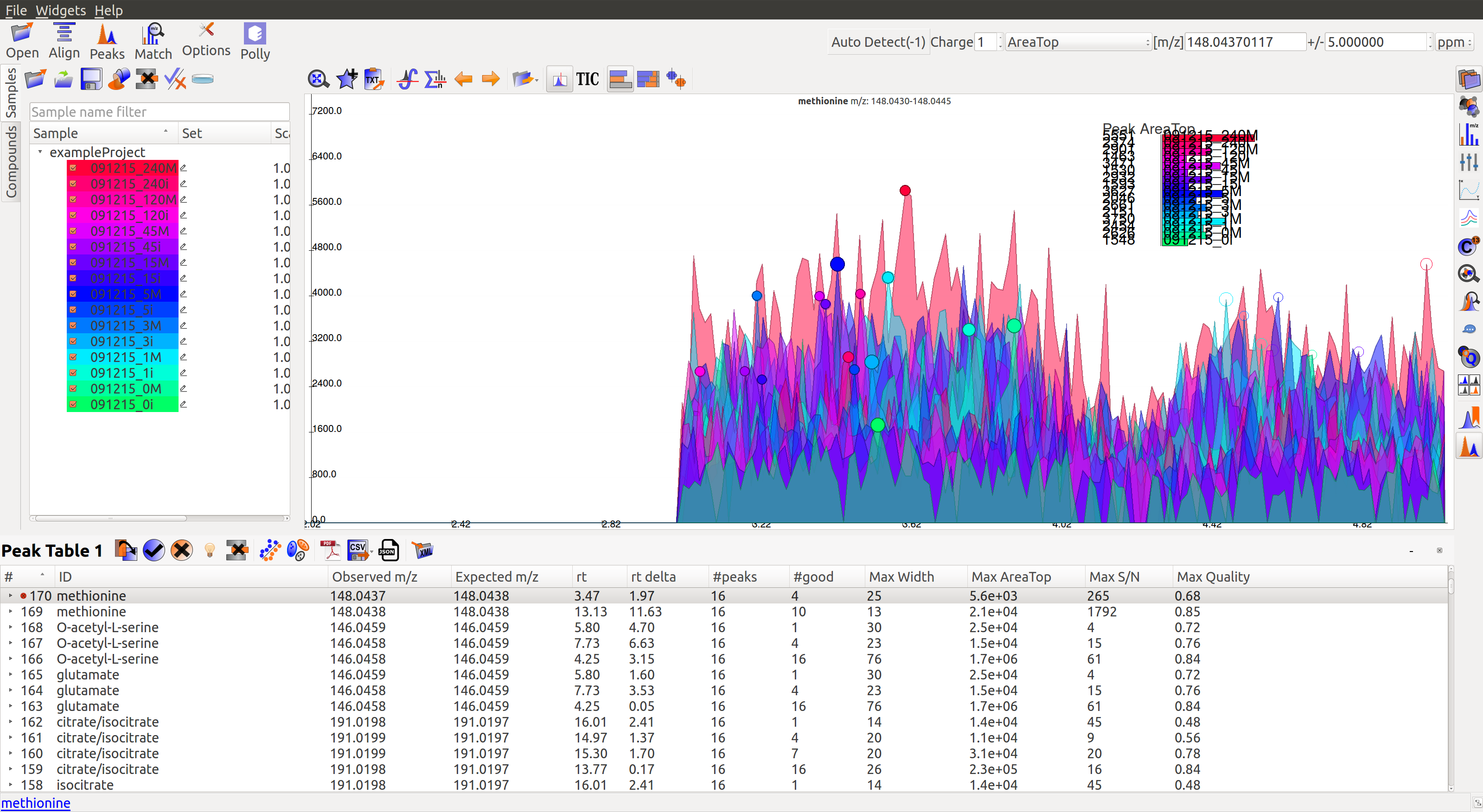
An Extracted Ion Chromatogram is a graph of Intensity vs. retention time for a certain m/z range. EIC window displays the EIC for every group/compound selected or m/z range provided. The group name and/or the m/z range is displayed at the top. Following are the different menu options on top of the EIC window:
Zoom Out: The EIC is initially zoomed-in to display the region near the expected retention time of a group. This button will zoom out and display the whole retention time range for the selected m/z range. Users may zoom in to a region by right dragging the mouse over it. Left-dragging will zoom out.
Copy Group Information to Clipboard: On clicking this button, group information is copied to the clipboard with every row representing a different sample.
Bookmark as Good Group: Users can manually curate a group as ‘good’ and store it in the bookmark table using this button. (Manual curation of groups has been covered here)
Bookmark as Bad Group: User can manually curate a group as ‘bad’ and store it in the bookmark table using this button. (Manual curation of groups has been covered here)
History Back: EIC window display history is recorded. Clicking this button will display the previous state of the window.
History Forward: EIC window display history is recorded. Clicking this button will display the next state of the window, if available.
Save EIC Image to PDF File: Saves the current EIC window display in a PDF file.
Copy EIC Image to Clipboard: Current EIC window display is copied to clipboard.
Print EIC: Current EIC window display can be directly printed out.
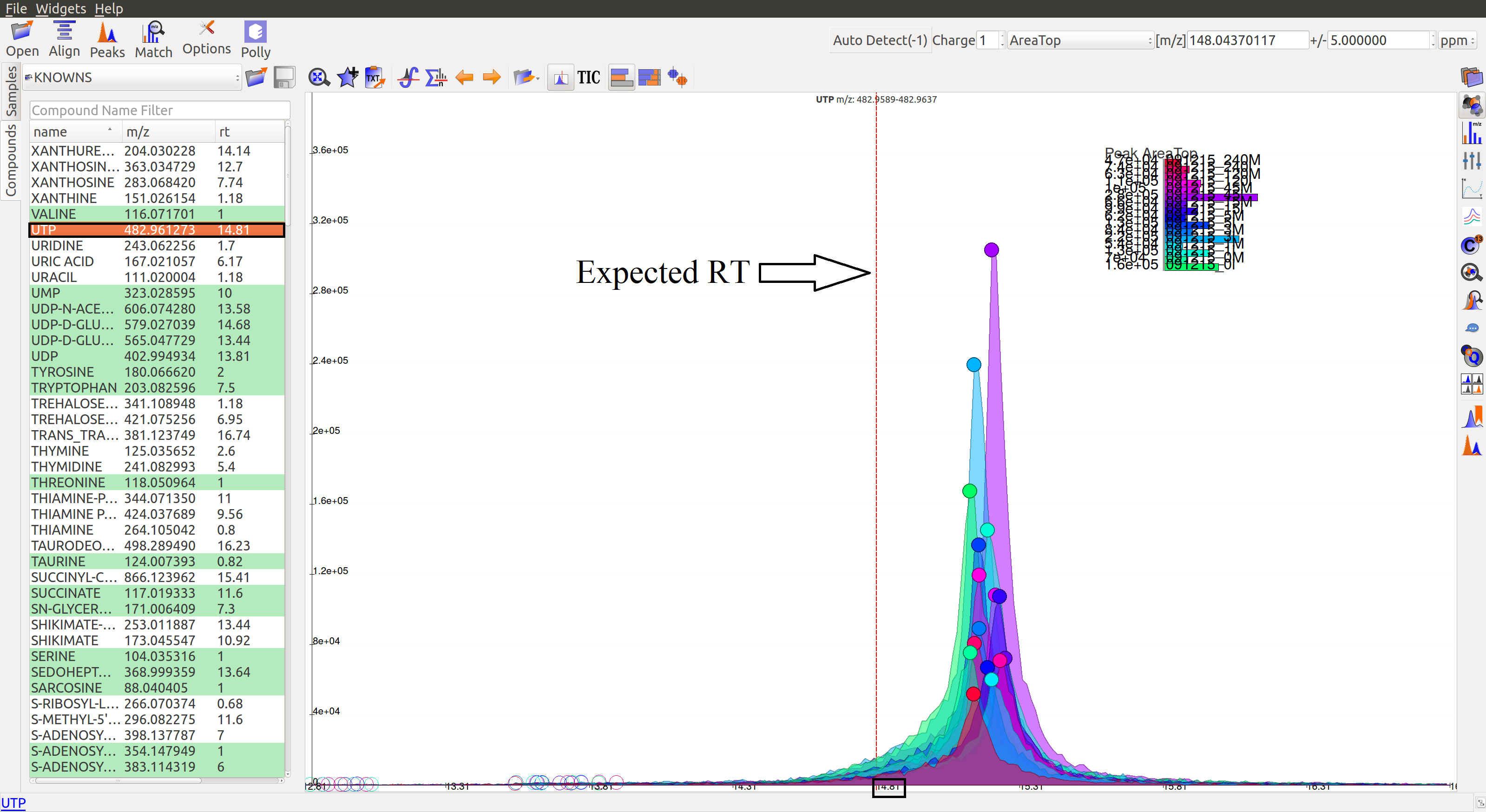
Auto Zoom: Auto Zoom is selected by default. It zooms-in and centers the EIC to the expected RT. The expected retention time is depicted as a dashed red line.
Show TICs: Displays the Total Ion Current. TIC is the sum of all intensities in a scan.
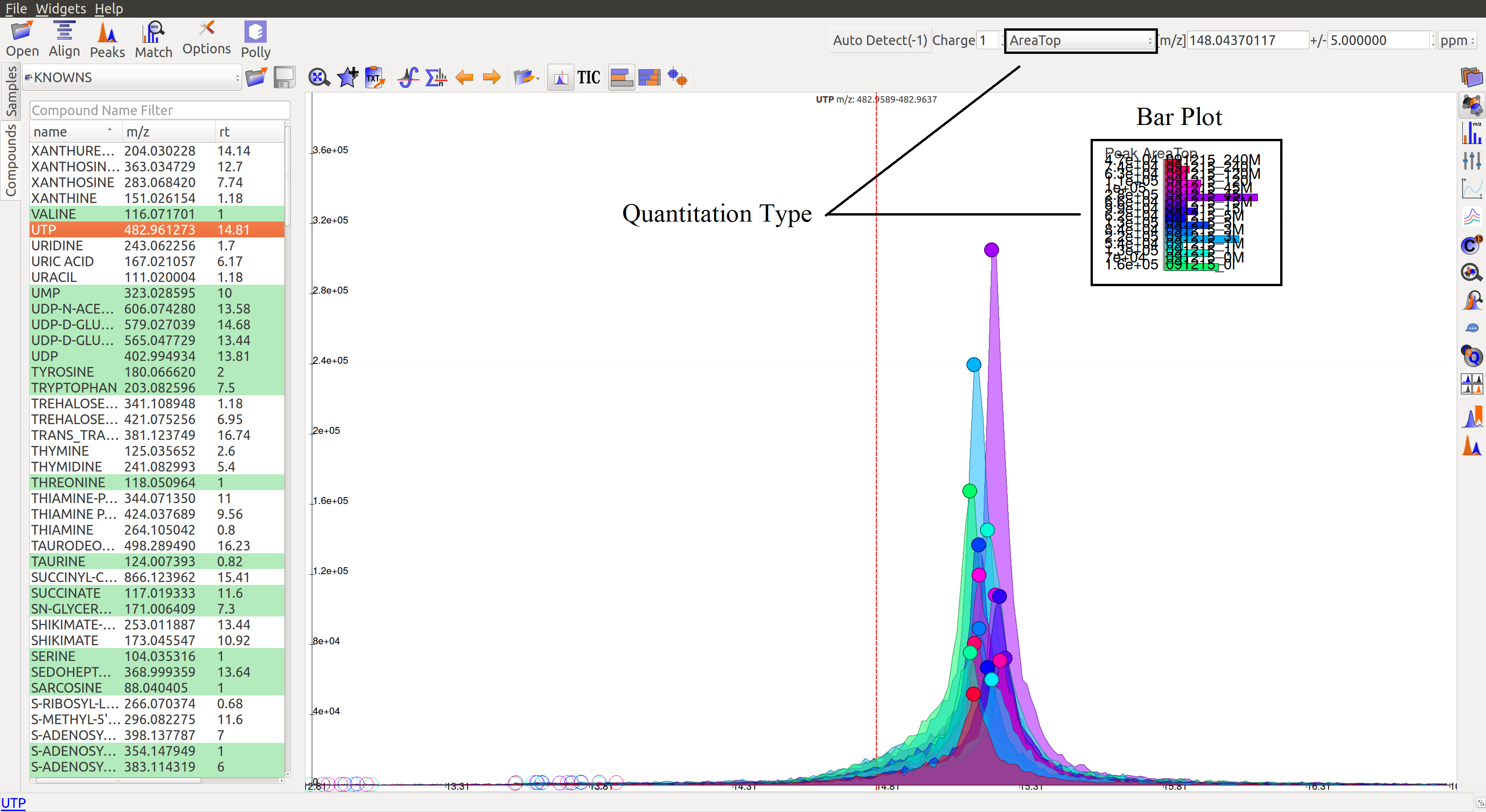
Show Bar Plot: Displays the peak intensity for a group in every sample. Intensity can be calculated by various methods known as quantitation types in El-MAVEN. Users can change the quantitation type from the drop-down list on the top right or choose to display other parameters like retention time and peak quality.
Show Isotope Plot: Displays the isotope plot for a group. Each bar in the plot represents the relative percentage of different isotopic species for the selected group in a sample.

Show Box Plot: Displays the boxplot for a group. The box plot shows the spread of intensities in the group and where each peak lies in relation to the median. Median of the intensities is the vertical line between the boxes.
Apart from the top menu, there are other features in the EIC window. Right-click anywhere in the window and go to Options.
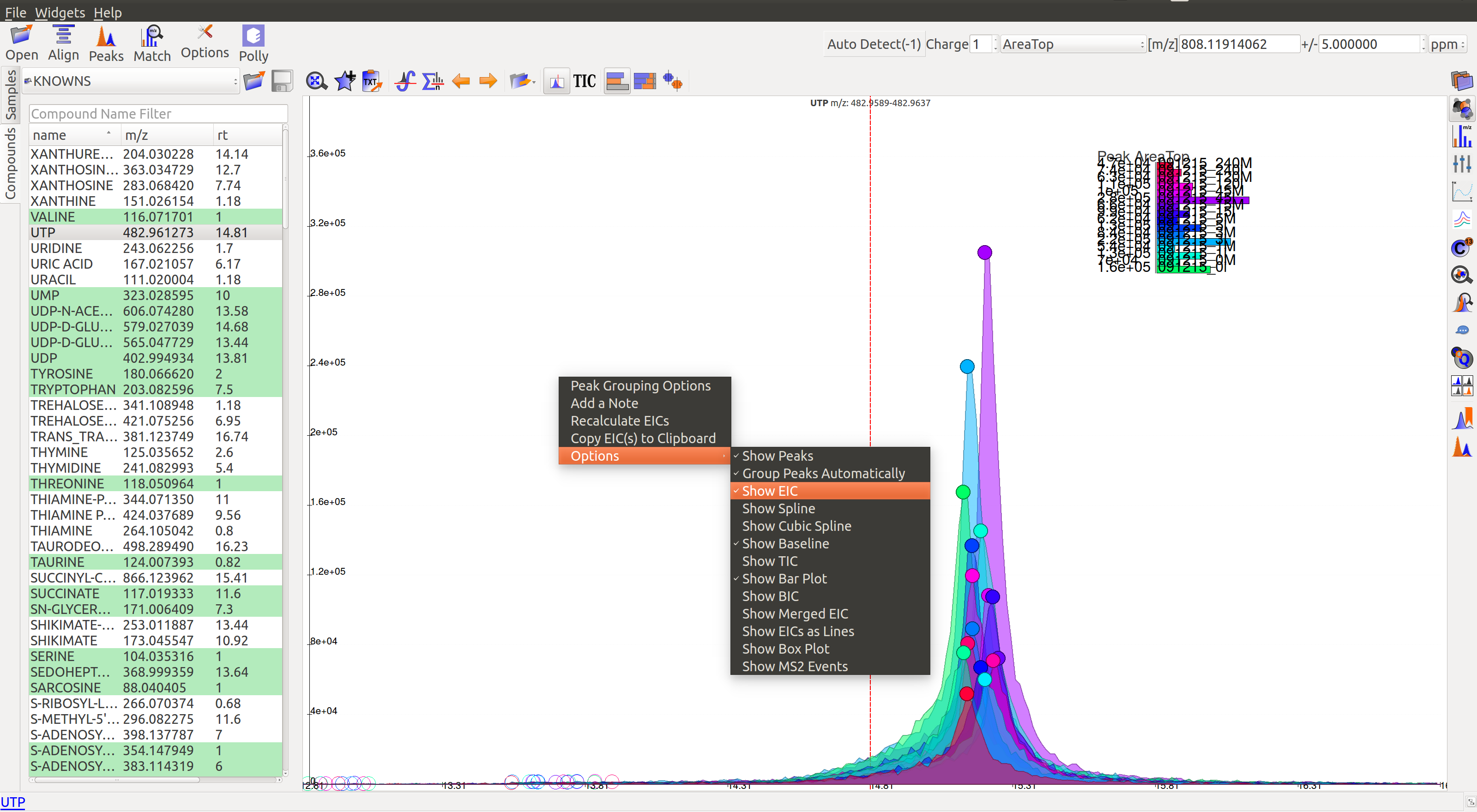
Some of the important options are:
- Show Peaks: Peaks are marked by the colored circles that represent the quality score of the peak. Bigger the circle, better the peak quality. This option allows the user to show/hide the peak quality score.
- Group Peaks Automatically: Peak grouping happens automatically when grouping parameters are changed. To prevent automatic grouping, user can uncheck this option.
- Show Baseline: Hide/Show the baseline for every peak. (Read more about baseline here).
- Show Merged EIC: Merged EIC is the sum average of EICs across samples. It smoothens the data and helps in grouping peaks.
- Show EIC as Lines: In case of large number of samples, it can get difficult to look at short individual peaks as they are obscured by larger peaks. Showing EIC as lines cleans up the display window and allows the user to look at small peaks.
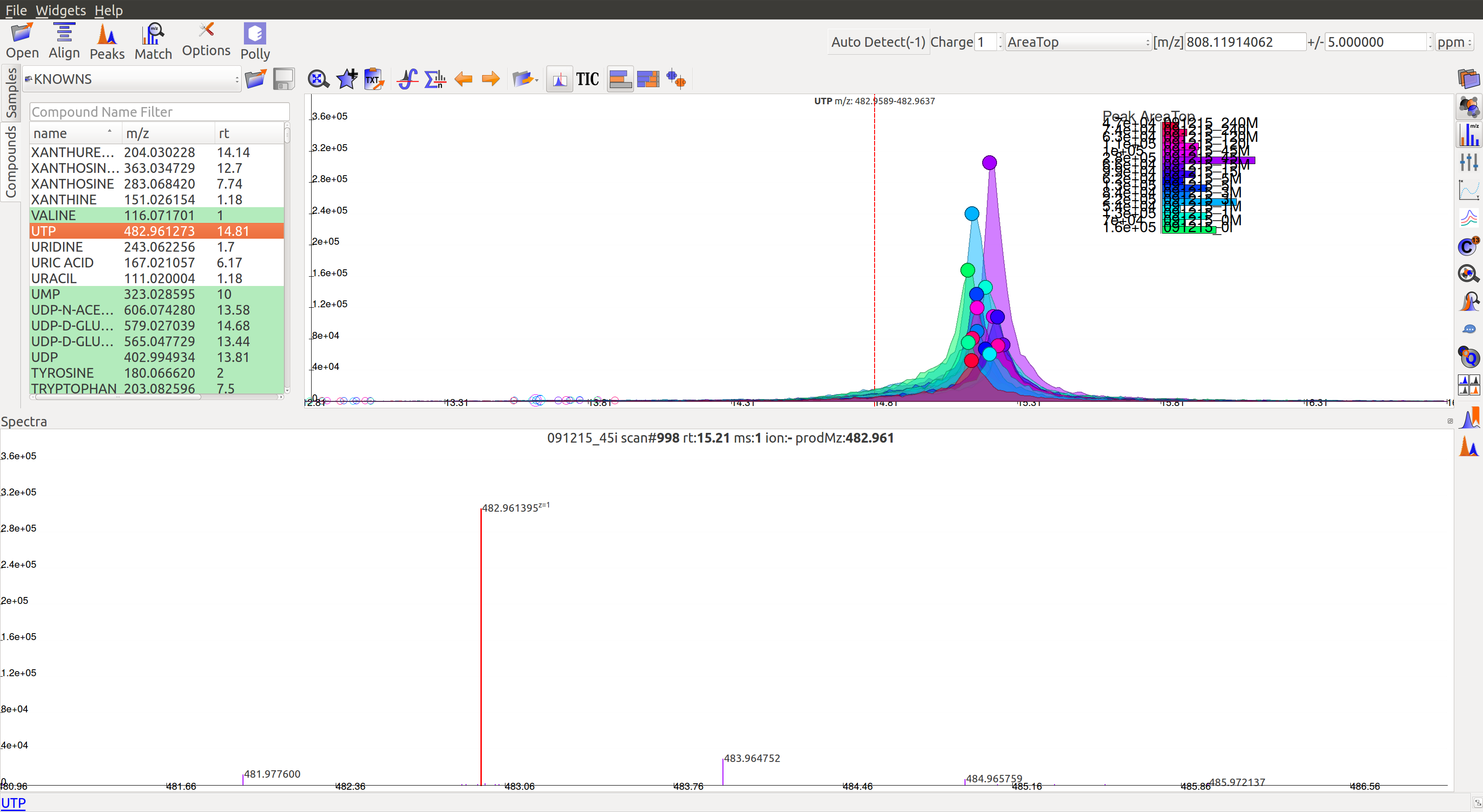
Mass Spectra¶
Mass Spectra Widget displays each peak, its mass, and intensity for a scan. As the widget shows all detected masses in a scan, the ppm window for the EIC and consequently grouping can be adjusted accordingly. This feature is especially useful for MS/MS data and isotopic detection.
Alignment¶
Prolonged use of the LC column can lead to a drift in retention time across samples. Alignment shifts the peak RTs in every sample to correct for this drift and brings the peaks closer to median retention time of the group.
Click on the Align button  and adjust the settings.
and adjust the settings.
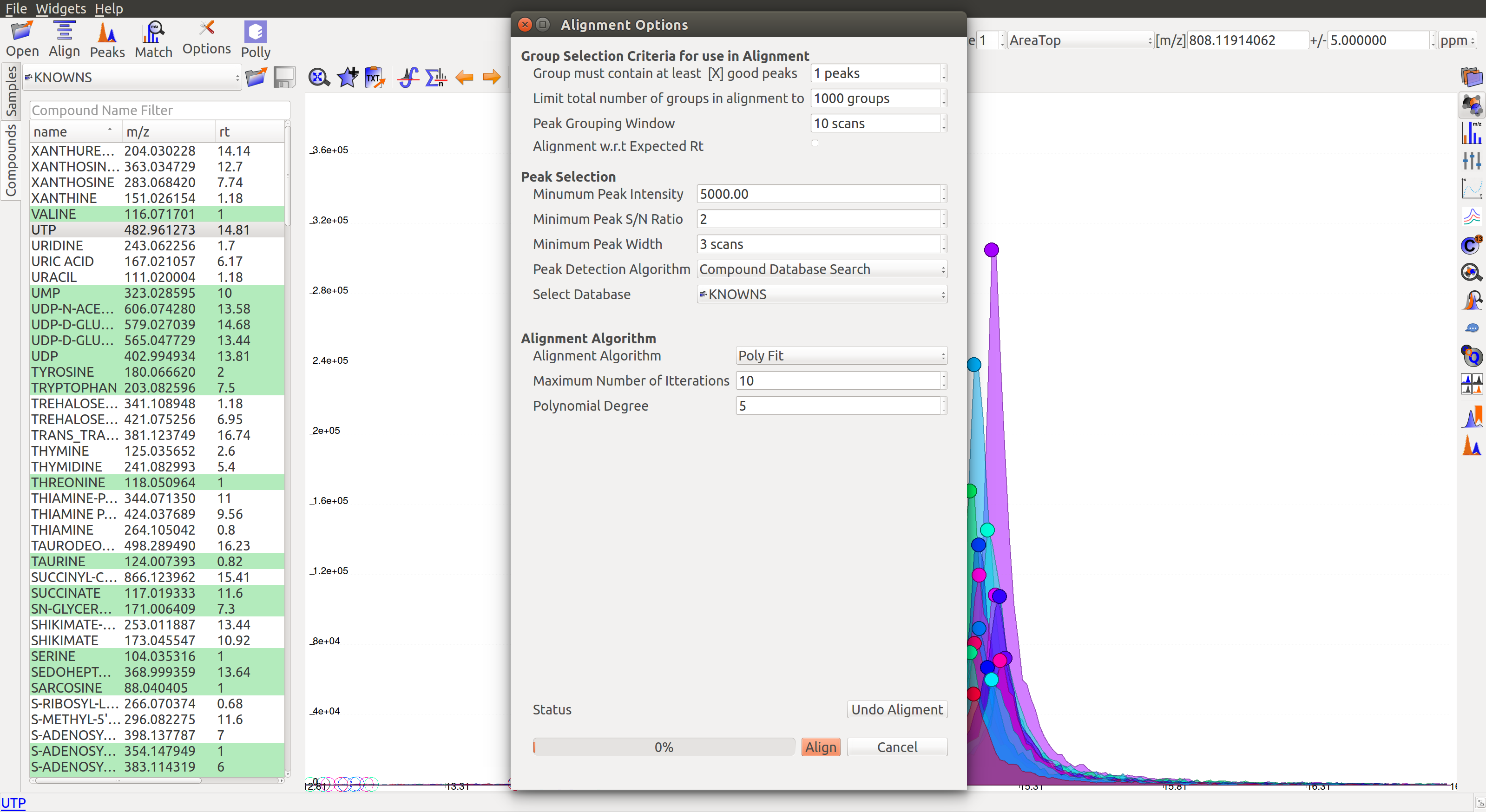
The first panel in Alignment options is for Group Selection criteria. ‘Group’ here refers to a set of peaks across samples that is annotated as a particular ion.
- Group must contain at least [X] good peaks: The value of x is set to filter out groups that do not have at least x good peaks from the alignment process. As there is only one peak per sample for a group, this value should not exceed the number of samples in your project. This option allows the users to discard groups with very few good peaks under the assumption that those could be stray peaks.
- Limit total number of groups in alignment to: Users can change the number of groups being used for alignment in case there are too many groups detected after the peak detection process.
- Peak Grouping Window: This value controls the number of scans required to get the most accurate peaks. Enter a high number if the reproducibility is low to ensure best results.
The next panel is for Peak Selection settings:
- Minimum Peak Intensity: The intensity value can be adjusted to only look at high or low intensity peaks in case you have prior information about the concentration of metabolite users are looking for.
- Minimum peak S/N ratio: This is the minimum signal to noise ratio of your experiment. Increase the value if you see too much noise in the data.
- Minimum Peak Width: This is the least number of scans to be considered to evaluate the width of any peak.
- Peak Detection Algorithm: Select the Compound Database Search algorithm and then choose an appropriate database from the next drop-down menu.
The Alignment Algorithm panel provides the following options:
- Alignment Algorithm: There are three alignment algorithms available in El-MAVEN: Obi-Warp, Poly fit and Loess fit. Loess fit has been released as a beta feature for now.
- Maximum number of Iterations: This parameter is only required for Poly fit algorithm. Enter the number of times El-MAVEN should fit a model to the data in order to align it.
- Polynomial Degree: This is the degree of the non-linear model we are trying to fit. Recommended settings are entered by default.
Click on Align at the bottom.
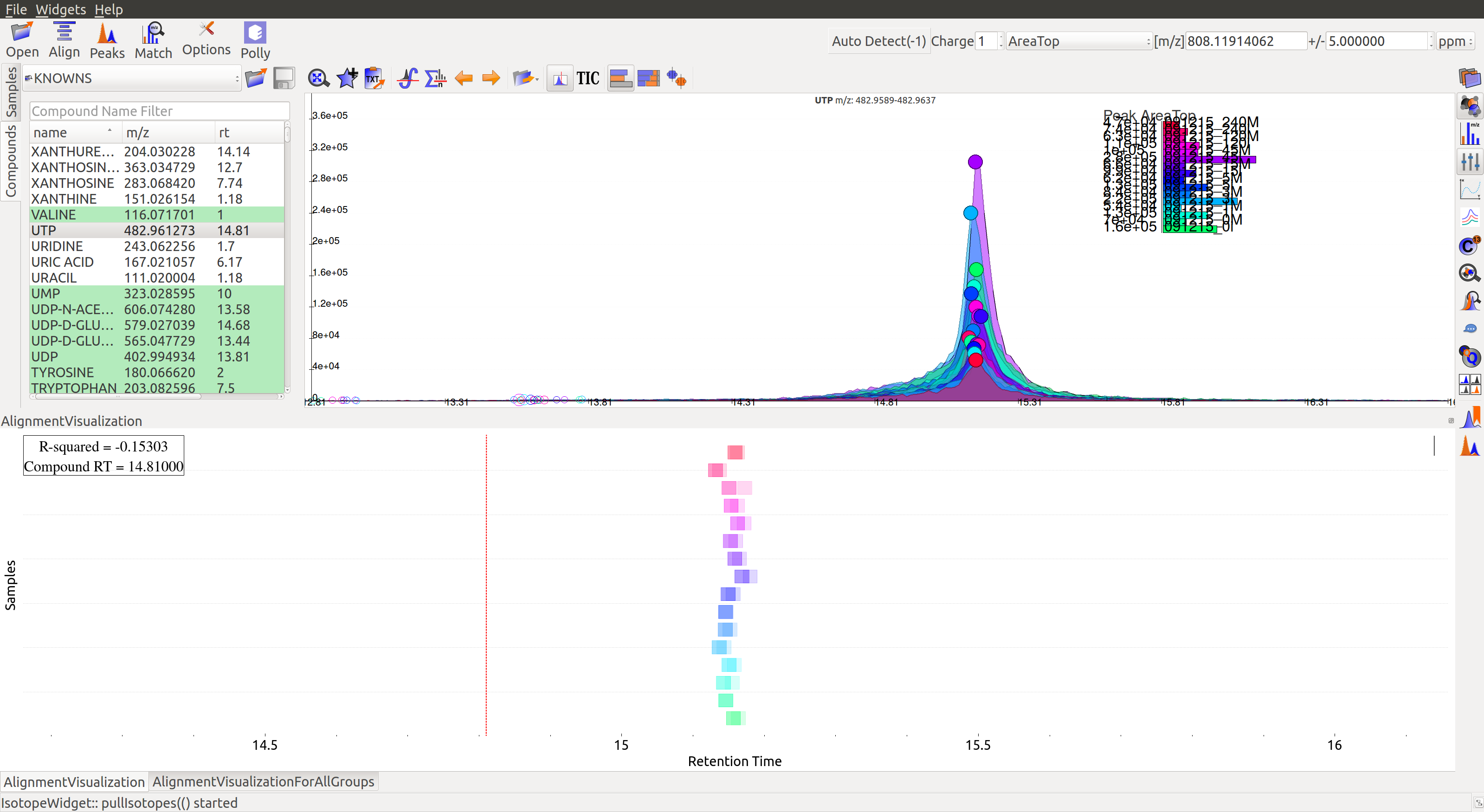
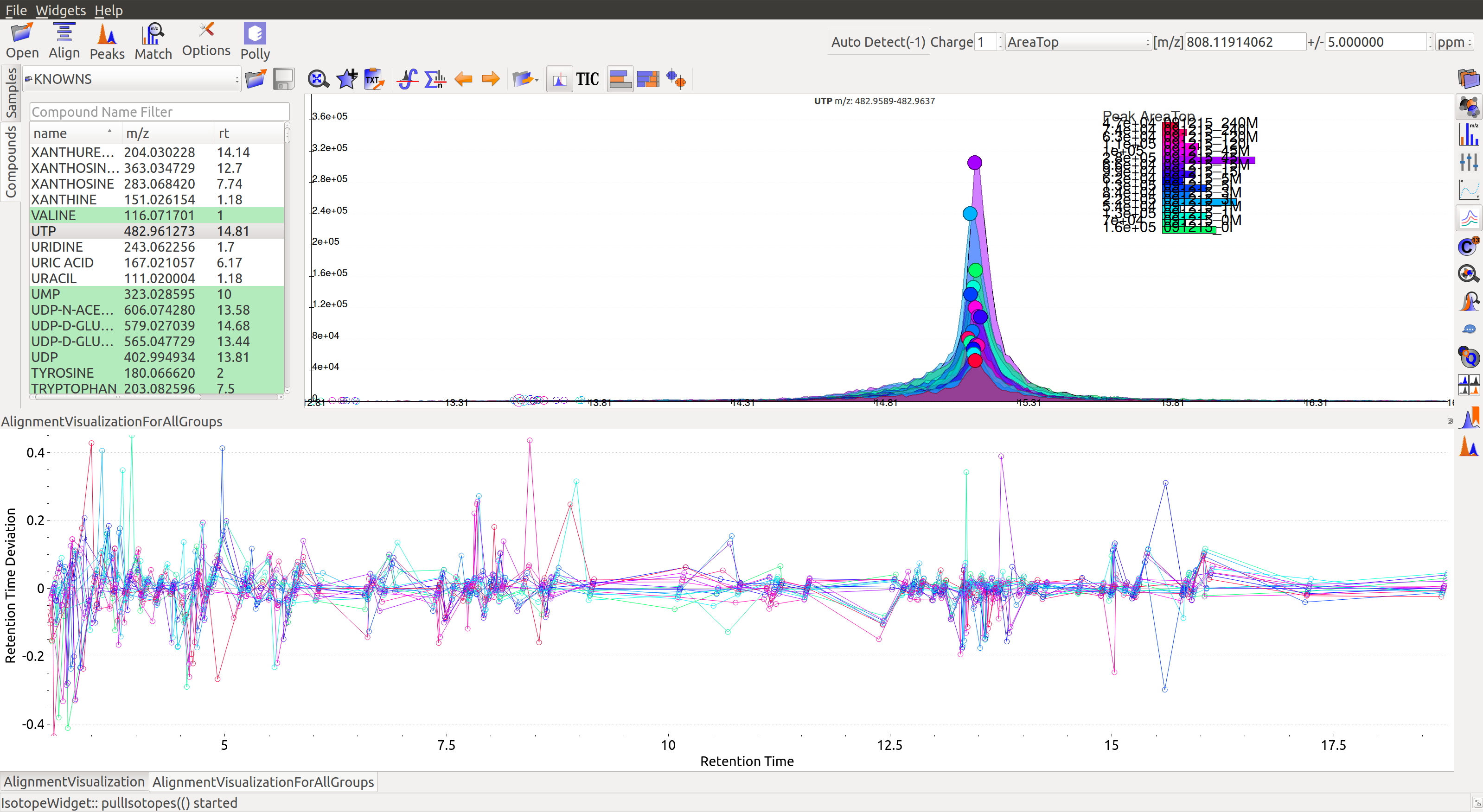
Alignment Visualizations
El-MAVEN provides three visualizations for alignment analysis.
- Show Alignment Visualization: Click on
 in the widget bar to open this visualization. Click on any grouped peak to look at its delta RT vs RT graph as shown.
in the widget bar to open this visualization. Click on any grouped peak to look at its delta RT vs RT graph as shown.
- Show Alignment Visualization (For All Groups): Click on
 in the widget bar for this visualization.
in the widget bar for this visualization.
- Show Alignment Polynomial Fit: Click on
 in the widget bar for Poly fit alignment.
in the widget bar for Poly fit alignment.
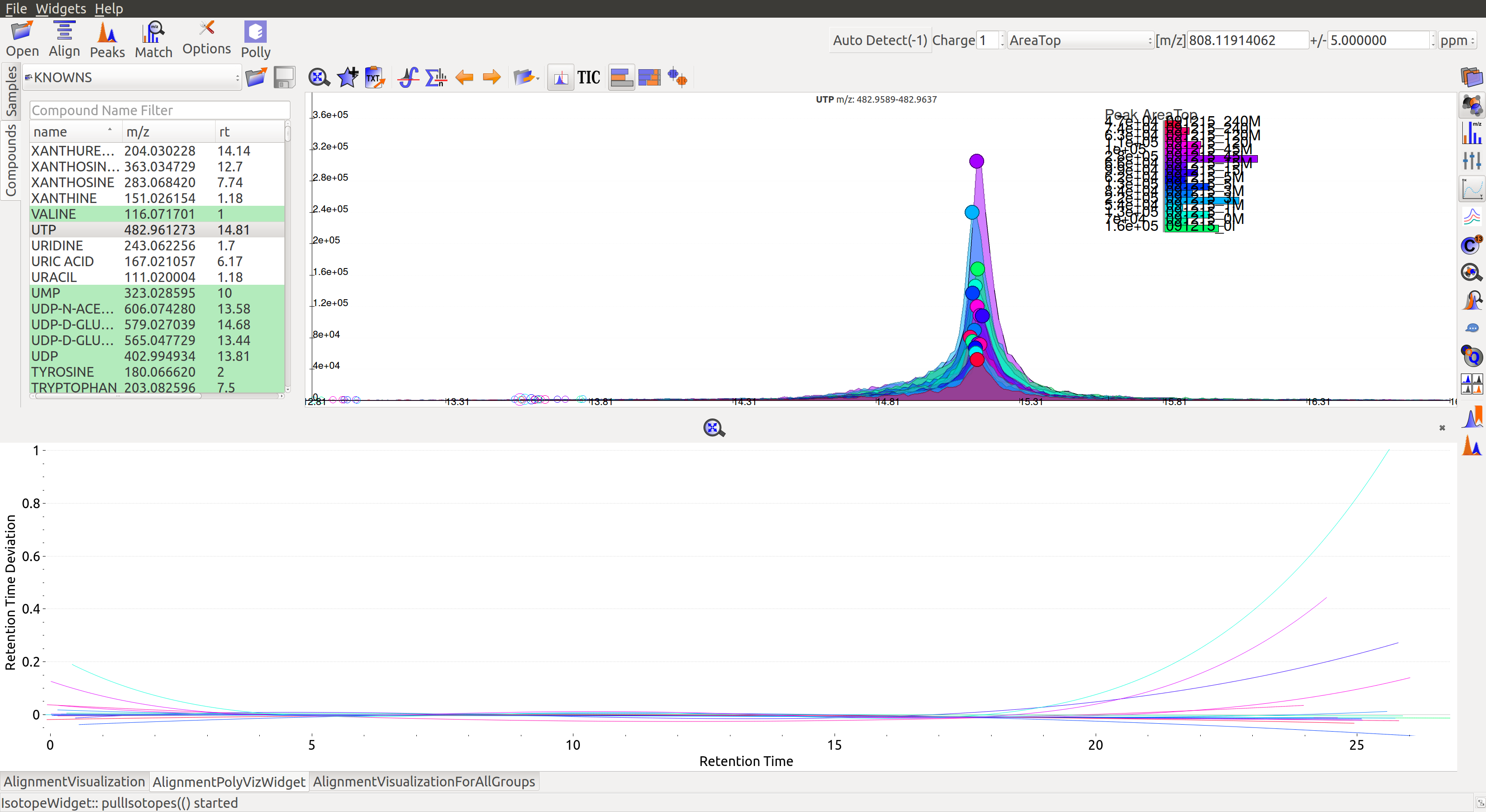
The above graphs give a clear indication of how aligned/misaligned the peaks are. Users may run alignment again with different parameters if required (or with a different algorithm).
Peak Detection¶
Peak detection algorithm pulls the EICs, detects peaks and performs grouping and filtering based on parameters controlled by the users. The algorithm groups identical peaks across samples and calculates the quality score by a machine learning algorithm. Click on the Peaks icon  on the top to open the settings dialog.
on the top to open the settings dialog.
There are 3 tabs for setting Peak Detection parameters:
1. Feature Detection Selection
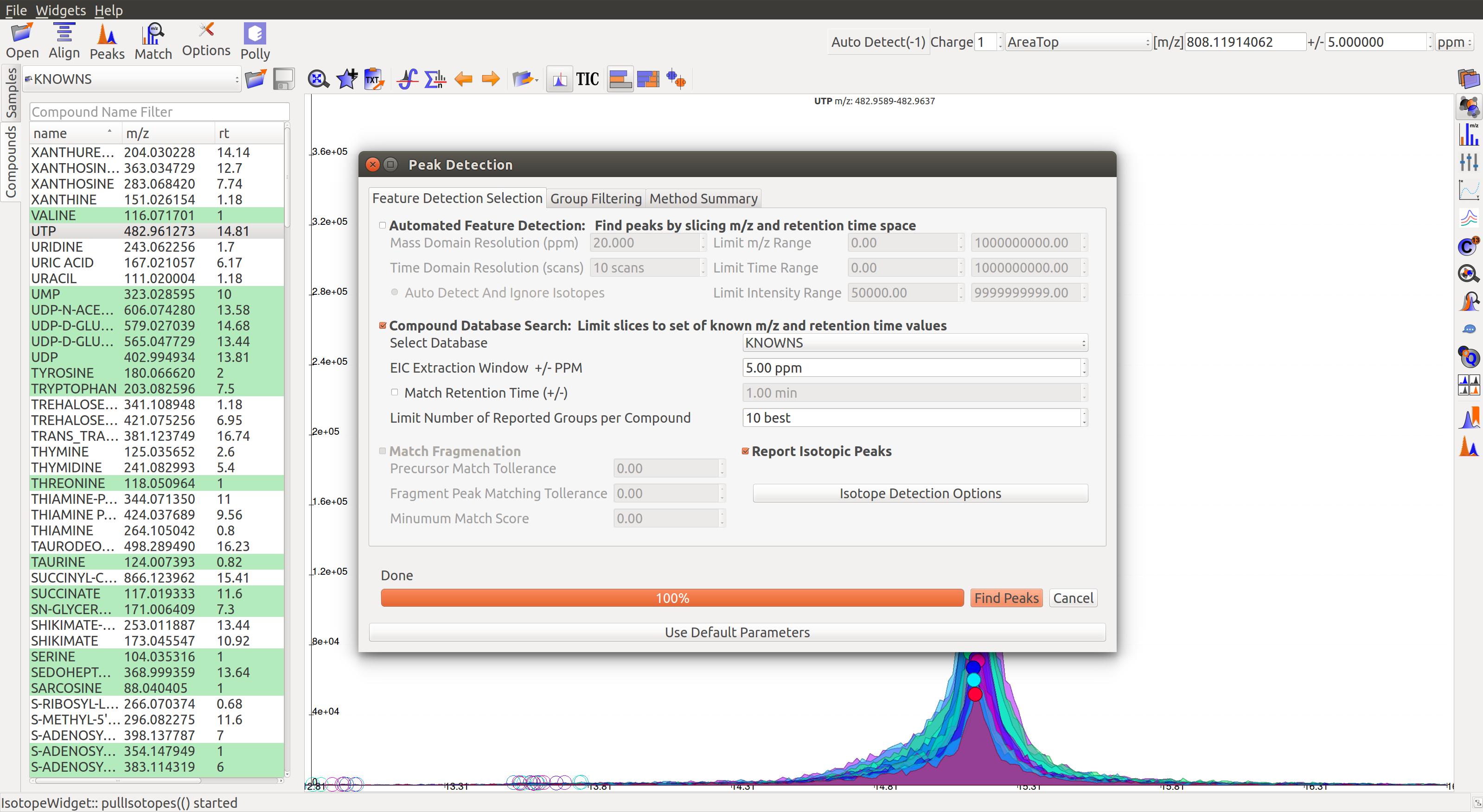
The Feature Detection Selection panel has the following parameters:
Automated Feature Detection: This is one of the two strategies for finding peaks. Automated search creates thousands of mass slices across the whole m/z and retention time space to find all peaks present in the sample. This strategy is used when looking for new/unknown metabolites in the samples.
- Mass Domain Resolution: This value defines the m/z range of every mass slice in parts per million
- Time Domain Resolution: This value defines the scan range (or retention time range) of every mass slice
- Limit Mass Range: User can limit the automated search to a range of m/z according to their requirements
- Limit Time Range: User can limit the automated search to a retention time range according to their requirements
Compound Database Search: Database search is used to search for compounds listed in the reference file using their m/z information. For better accuracy, retention time information can also be used for the search.
- Select Database: Select a desired reference file for the search from the drop-down list
- EIC Extraction Window: Provide a ppm buffer range to all compound masses. A larger window is useful for processing low resolution data. The window should be smaller for high resolution data to reduce noise.
- Match Retention Time: Enable/disable use of retention time information along with m/z to perform database search. Compounds can have different retention times in every experiment, therefore this option should only be checked if the reference file is specific to the experiment and the sampled used. Enter the time buffer in the accompanying box.
- Limit Number of Reported Groups Per Compound: Multiple groups can be annotated as the same compound, especially when retention time is not taken into consideration for the search. Users can set the value to only report X best groups according to their rank. The group rank formula will be discussed later in the tutorial.
Match Fragmentation: This panel is activated for MS/MS data.
Report Isotopic Peaks: Check this box to find and report isotopic peaks for labeled data.
To perform peak detection with reference, check the box next to Compound Database Search and choose the appropriate database. The EIC Extraction Window should be set according to the instrument’s resolving power. Select the Match Retention Time option if you wish to search for compounds using both the m/z ratio and retention time value given in the database. In case of a generic database, searching by retention time is not recommended.
2. Group Filtering

After grouping is done, groups that do not fulfill the criteria shown above are filtered out.
- Minimum Peak Intensity: Groups with no peak intensities above this threshold are filtered out. The drop-down list beside the input box defines how intensity is calculated. Different methods of intensity calculation are known as quantitation types. The slider below can be adjusted to change the minimum percentage of peaks per group that must pass the threshold (minimum number of peaks is 1).
- Minimum Quality: Quality of peaks is calculated using a machine learning algorithm. Groups with no peak qualities above this threshold are filtered out. The slider below can be adjusted to change the minimum percentage of peaks per group that must pass the threshold (minimum number of peaks is 1).
- Minimum Signal/Blank Ratio: Signal/Blank ratio is the ratio of peak intensity over maximum intensity observed in blanks. Groups with no peaks above this threshold are filtered out. The slider can be adjusted to change the minimum percentage of peaks per group that must pass the threshold (minimum number of peaks is 1). This helps in filtering out peaks that are also present in blanks.
- Minimum Signal/Baseline Ratio: Signal/Baseline ratio is the ratio of peak intensity over baseline value for that peak. Baseline calculation is used to filter out noise in the signal and will be discussed later in the tutorial. The slider can be adjusted to change the minimum percentage of peaks per group that must pass the threshold (minimum number of peaks is 1).
- Minimum Peak Width: Peak width is equal to the number of scans that a peak is spread over. Groups with no peak widths above this threshold are filtered out. Spurious signals can be filtered out using this option.
- Peak Classifier Model File: This is the default model that is used by the machine learning algorithm for classifying peaks according to their quality.
Change the settings according to the data and click on Find Peaks to run peak detection. For beginners, performing peak detection with default values at first is recommended. Users may then adjust the settings depending on their results.
3. Method Summary
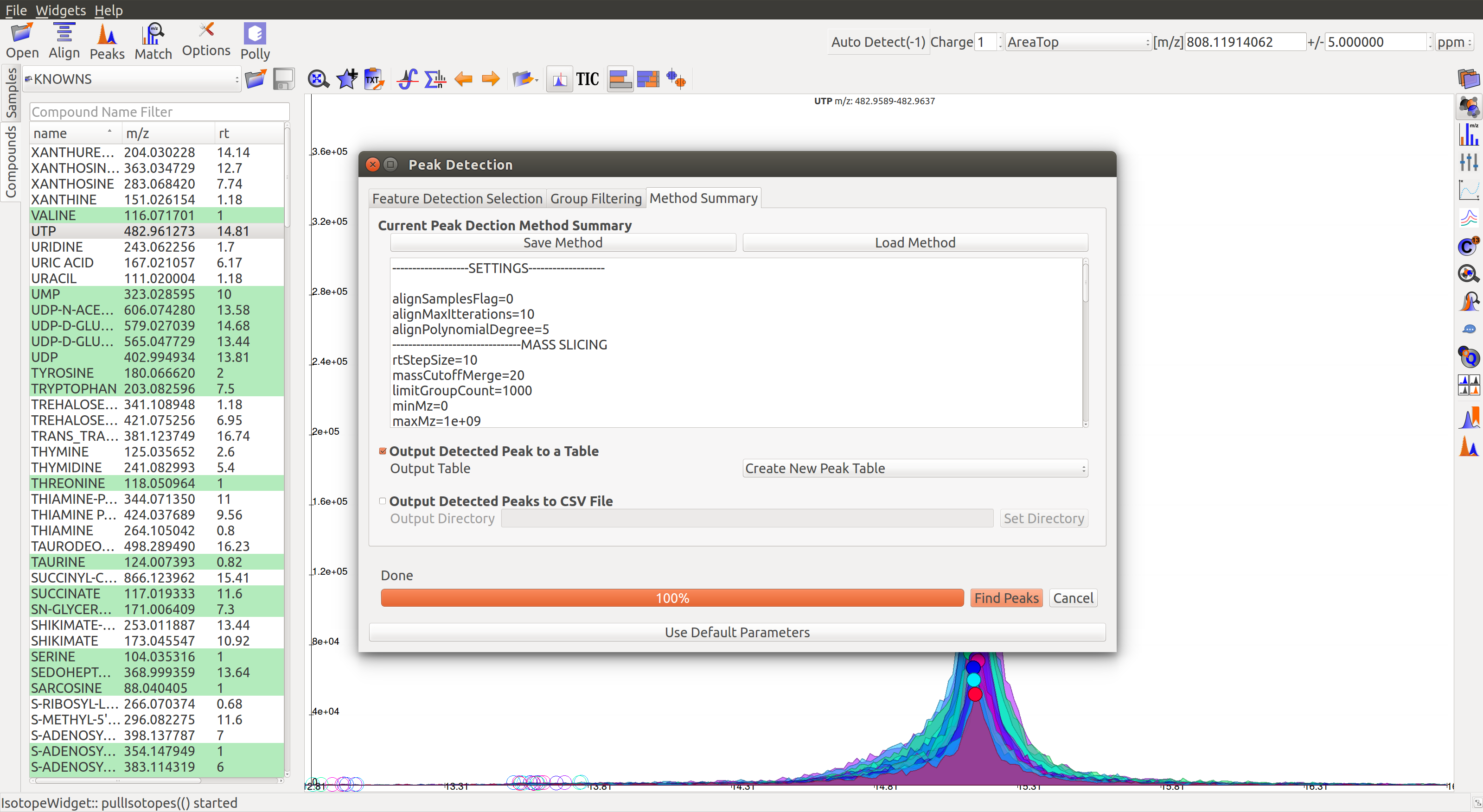
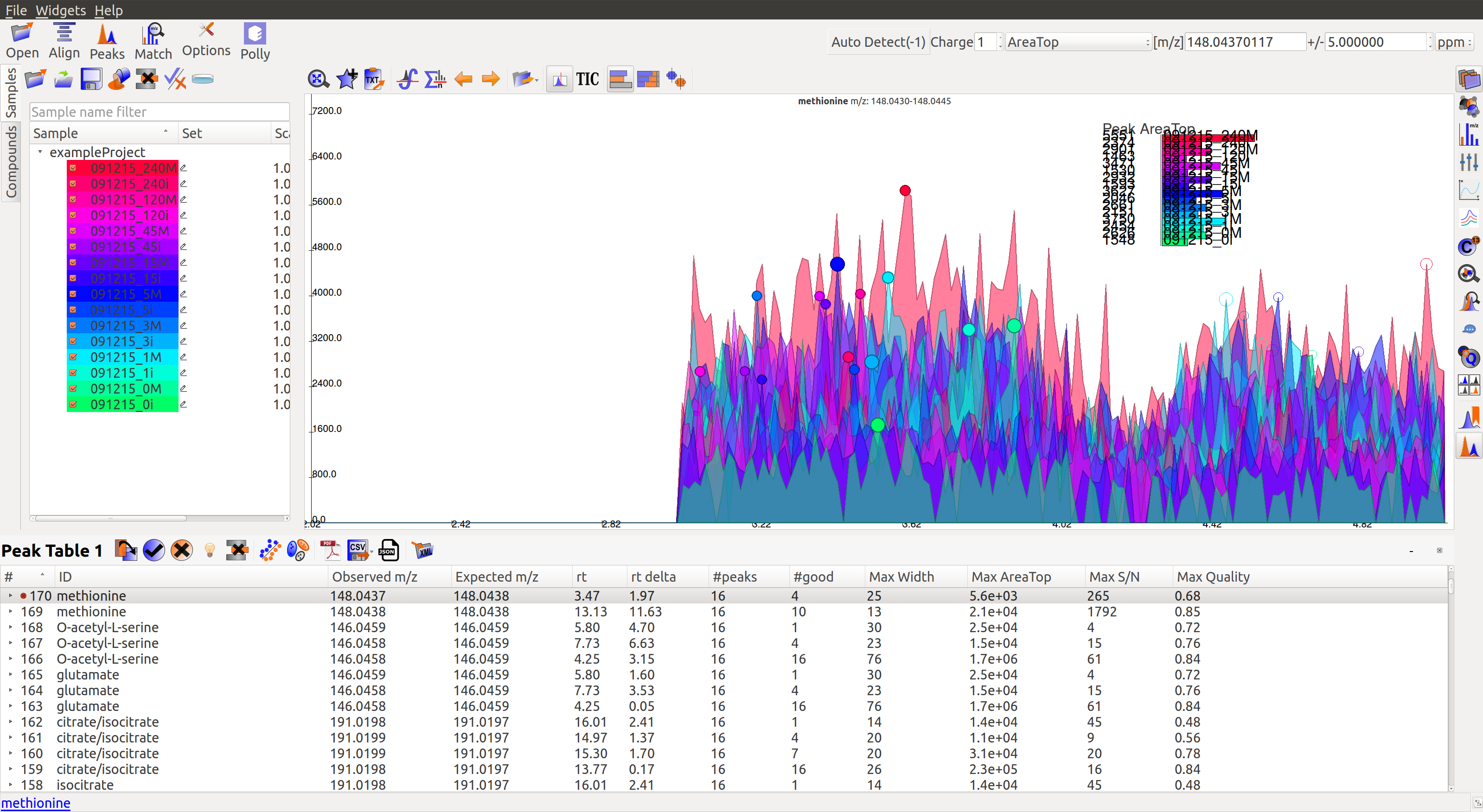
Peak Table¶
Groups information obtained after Peak Detection is stored and displayed in the form of a Peak Table with a row representing a group and its corresponding features in columns. Users can show/hide the peak table by clicking on the widget bar.
Peak Table Features
Following are the different features/columns in a peak table:
- #: is the serial number for a group
- ID: Group ID is assigned according to the search mode used during peak detection. In case of Automated search, groups are named by their m/z and retention time values separated by ‘@’ sign. For example, ID for a group with 230.2 m/z and 1.89 RT will be given as ‘230.2@1.89’. In case of Database search, groups are annotated as a compound from the reference file. For example, ‘malate’.
- Observed m/z: is the median m/z of the group.
- Expected m/z: is the m/z value provided in the reference file for the compound represented by the group. This field is populated only in case of Database search.
- rt: is the median retention time of the group.
- rt delta: is the difference between expected retention time from the reference file and the observed RT. This field is set to -1 in case of Automated Search.
- #peaks: is the number of peaks in the group.
- #good: is the number of good peaks in a group. A good peak is defined as one with its quality score above the defined threshold in Peak Detection dialog.
- Max Width: is the maximum peak width in a group. Peak width is defined as the number of scans over which a peak is spread.
- Max AreaTop: is the maximum peak AreaTop intensity in a group. AreaTop is one of the quantitation types used to represent peak intensity in El-MAVEN. Read more about the different quantitation types here.
- Max S/N: is the maximum peak signal/noise ratio in a group.
- Max Quality: is the maximum peak quality score in a group.
- Rank: is the group rank. The formula and parameters involved have been explained here.
Peak Table Menu Bar
Multiple groups can be annotated as the same compound especially when retention time information is not used during Database search. The peak table provides options for filtering, comparing or exporting data from the table. Following are the different menu options available in the peak table:
Switch between group and peak views: Switching to Peak view displays only Peak information. This includes group number, group ID, Expected m/z, Observed m/z, retention time and intensity of all peaks in the group with sample names as the respective column headers. Peak intensity cells are colored based on their relative values in a group. Highest intensity value has the lightest color and vice-versa.
Train Neural Net: Used to retrain the neural net algorithm to recognize good/bad peaks. User manually curates 100 peaks to train the algorithm.
Show Scatter Plot: Opens the Scatter plot widget used to compare different cohorts via Scatter plot and Volcano plot.
The remaining are export options and will be detailed in the Export section.
Statistics¶
El-MAVEN comes equipped with a statistics module for comparing data across different cohorts. Users can set the sample cohorts either by editing the Set column in the Sample space, or upload a meta file with sample and cohort names as detailed above under the Sample Space Menu section.
The statistics module can be accessed through the Peak Table menu.
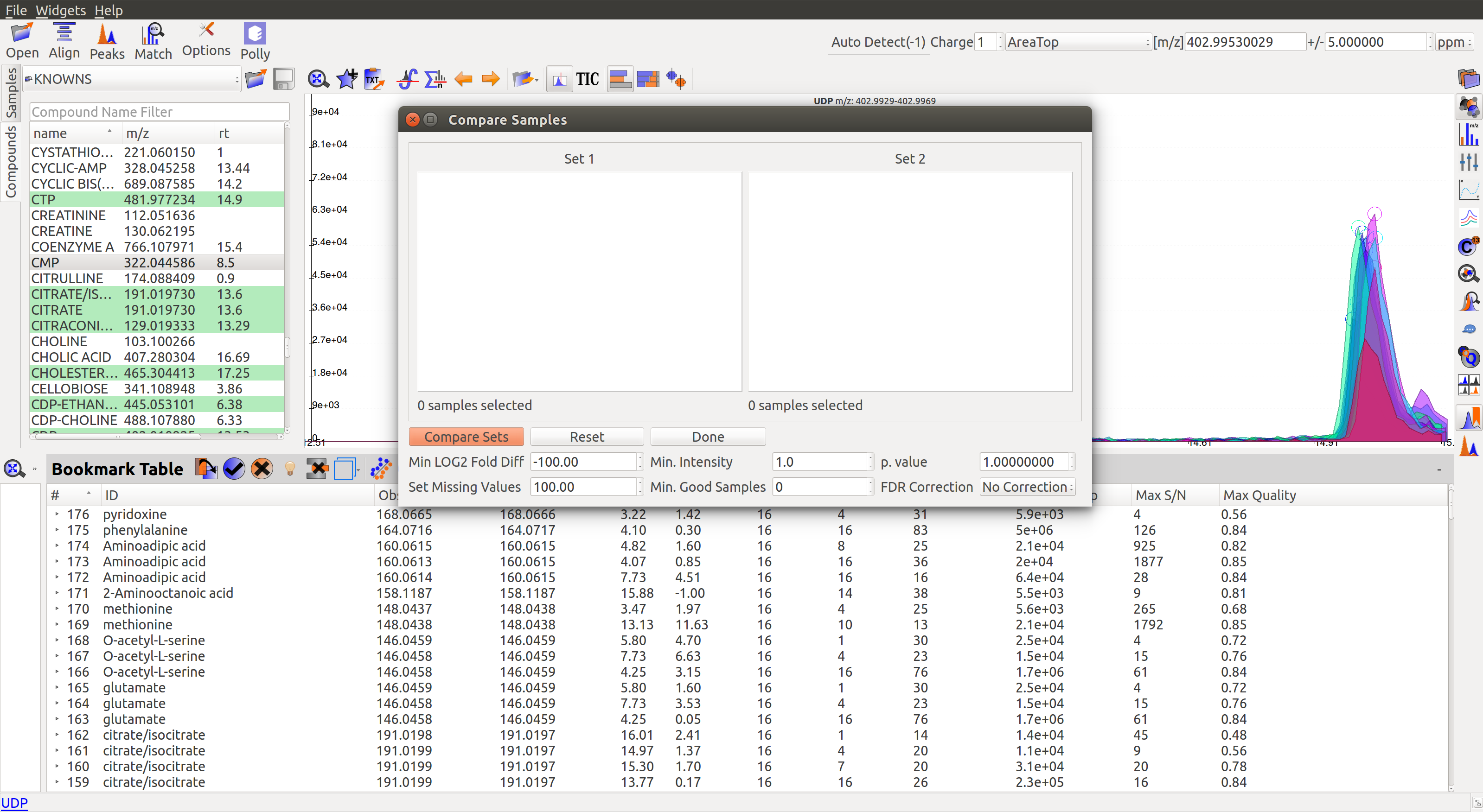
- Set1/Set2: Select two cohorts to be compared
- Min Log2 Fold Difference: Fold difference is a measure of how much the intensity of a group changes from one cohort to another. User can set the minimum threshold for this value in log2format.
- Min Intensity: Groups with all peak intensities less than this value will be filtered out from the comparison process.
- p value: A t-test is performed to find if the intensity distributions of the two selected cohorts are significantly different from each other. This test returns a p-value indicating how significantly different a group behaves between the two cohorts. A lower p-value shows higher significance.
- Set Missing Values: User can set the default intensity value to be used in case the group is missing from a sample.
- Min. Good Sample: Groups should have a minimum number of good peaks (based on peak quality score) to be considered for comparison.
- FDR Correction: False discovery rate is the expected proportion of false positives in a test. There are a number of ways to correct for false positives. (Read More)
- Compare Sets: Click to get comparison results.
Compare Sets opens the scatter plot by default
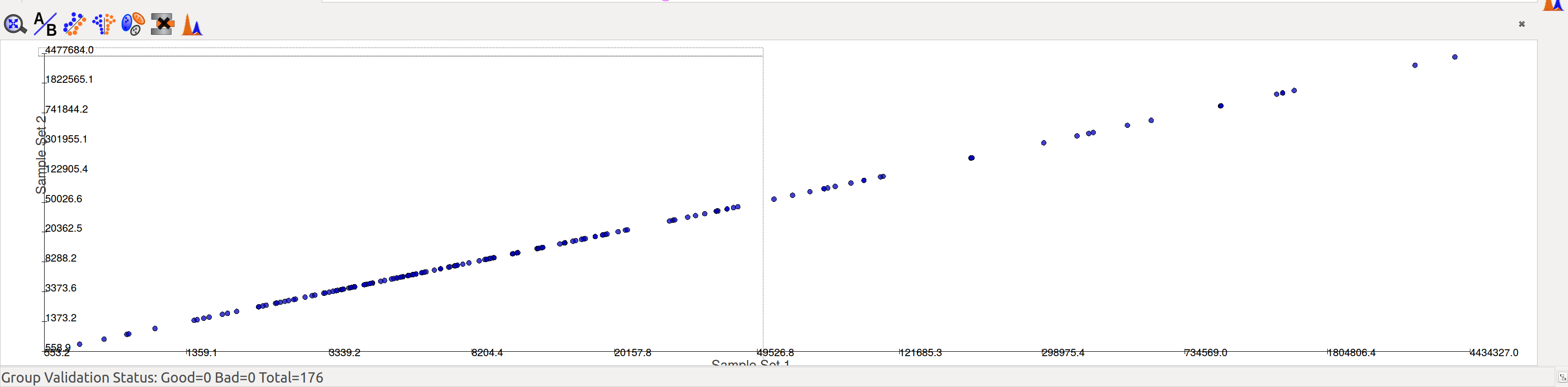
Compare Samples: Opens the compare samples dialog again to adjust settings.
Volcano Plot: The axes represent fold change and significance of fold change respectively. Red and blue bubbles represent positive and negative fold change respectively.
Export¶
Users can either save the state of the project or export only relevant data from the peak table. These are the different export options available in El-MAVEN:
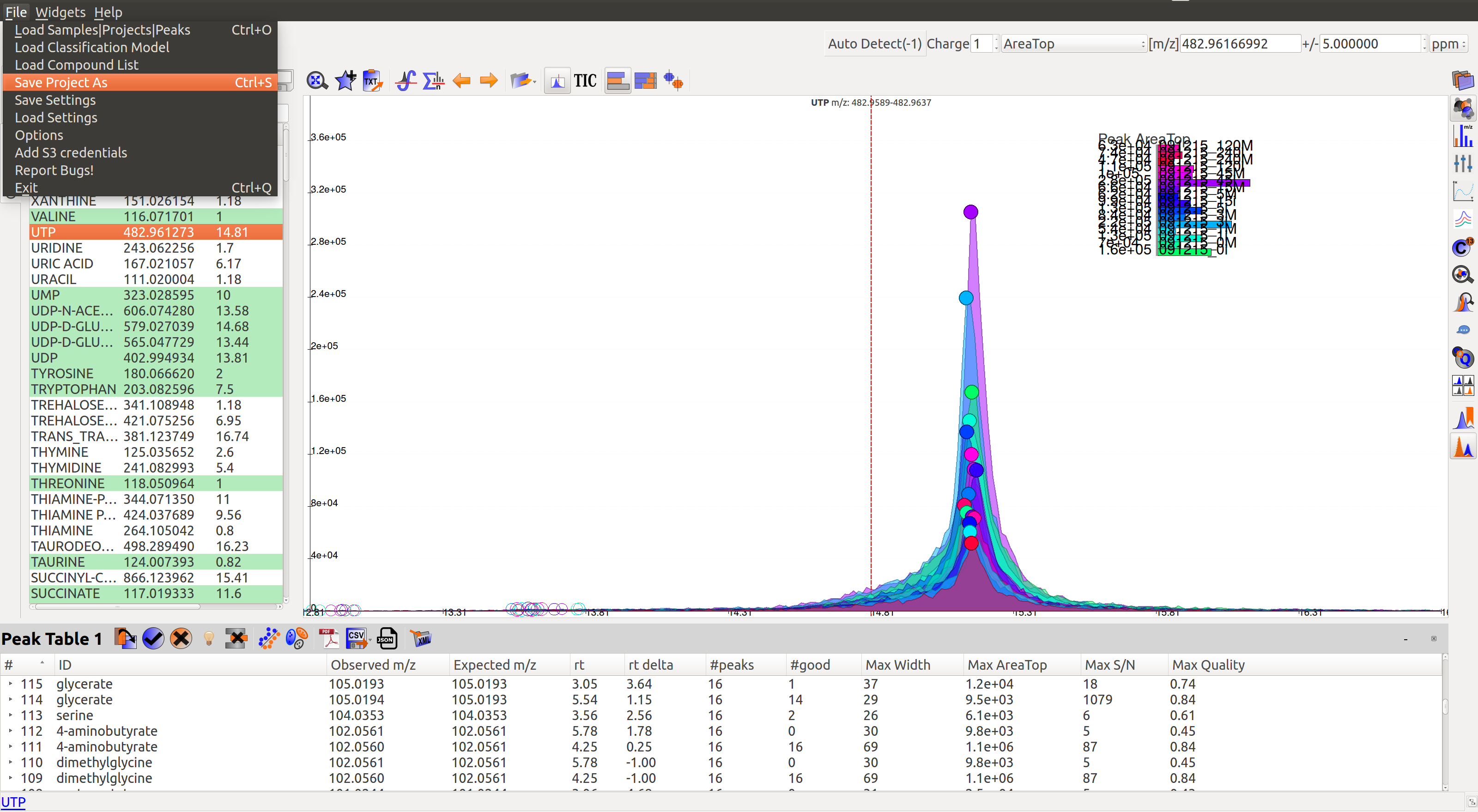
- Save Project as: This option is available in the File menu. It saves all peak tables and current settings in a .mzroll file. On loading the .mzroll file, all sample files are uploaded and the peak tables and EIC are available. If the user wishes to save only certain Peak Tables, they can click on
- Generate PDF Report: This option is available on
at the top of the Peak Table. It saves all EICs with their corresponding bar plots in a PDF file.
- Export Groups to SpreadSheet (.csv): This option is available on top of the Peak Table
. You can choose to export the whole table or a subset of the data. There are 4 possible selections: export only selected groups, export all groups, export only good groups or export only bad groups. The data is stored in a comma separated file.
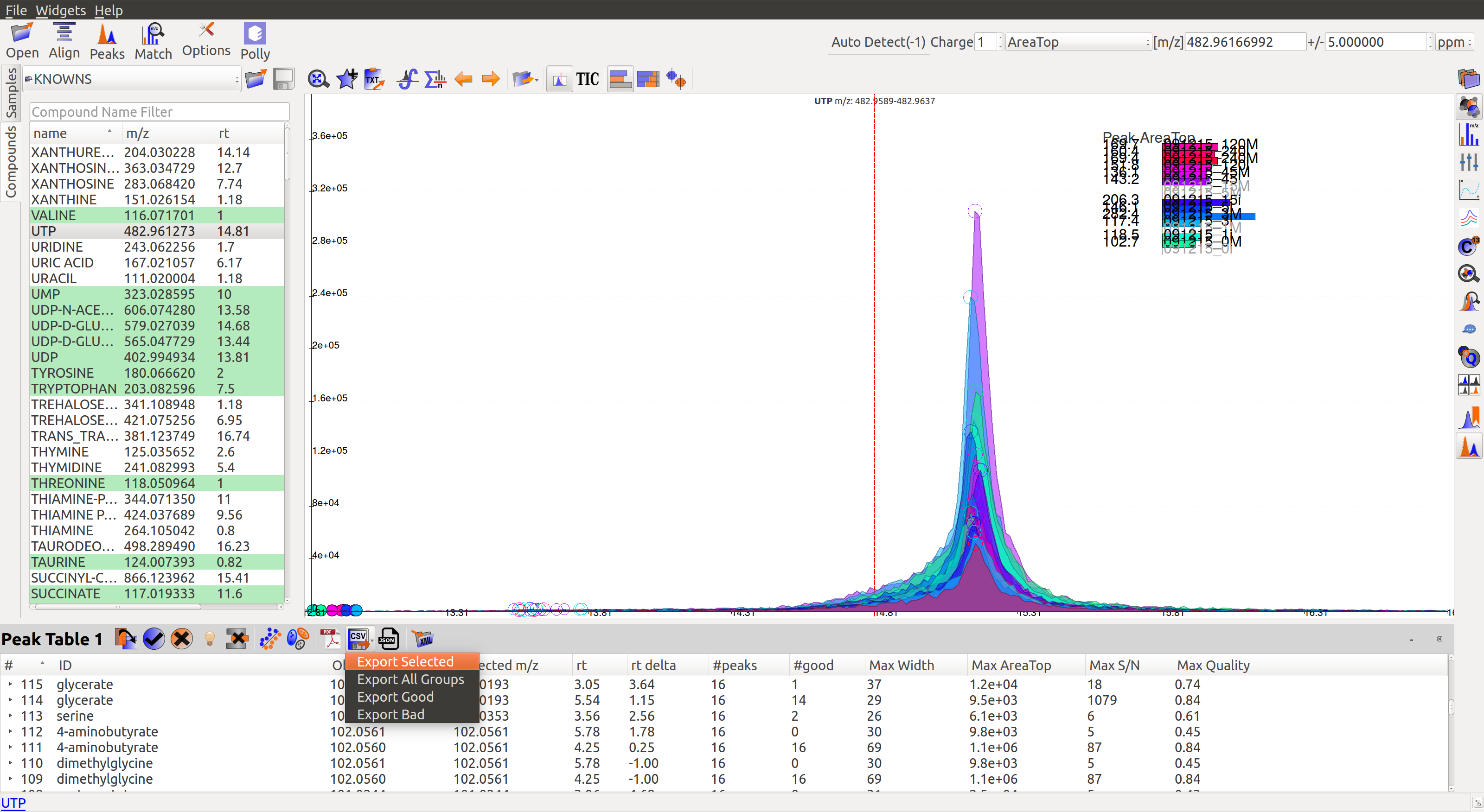
- Export EICs to Json: This option is available on top of the Peak Table
. It exports all EICs to a Json file.
